Pyridin
Pyridin ist eine farblose und leichtentzündliche chemische Verbindung mit der Summenformel C5H5N. Sie gehört zu den heterocyclischen Stammsystemen und bildet das einfachste Azin, das aus einem sechsgliedrigen Ring mit fünf Kohlenstoffatomen und einem Stickstoffatom besteht. Die Bezeichnung Azine leitet sich aus der systematischen Hantzsch-Widman-Nomenklatur ab, nach der Pyridin als Azin bezeichnet wird. In Analogie zu Benzol ist auch die Bezeichnung Azabenzol gelegentlich anzutreffen. Im Jahre 1849 wurde Pyridin erstmals von dem schottischen Chemiker und Mediziner Thomas Anderson beschrieben, der die Inhaltsstoffe von Knochenöl untersuchte. Zwei Jahre später isolierte Anderson Pyridin durch fraktionierende Destillation des Öls erstmals in reiner Form.
| Strukturformel | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
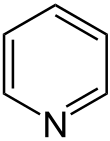 | ||||||||||||||||
| Allgemeines | ||||||||||||||||
| Name | Pyridin | |||||||||||||||
| Andere Namen |
| |||||||||||||||
| Summenformel | C5H5N | |||||||||||||||
| Kurzbeschreibung |
farblose, hygroskopische Flüssigkeit mit unangenehmem, charakteristischem Geruch[1] | |||||||||||||||
| Externe Identifikatoren/Datenbanken | ||||||||||||||||
| ||||||||||||||||
| Eigenschaften | ||||||||||||||||
| Molare Masse | 79,10 g·mol−1 | |||||||||||||||
| Aggregatzustand |
flüssig | |||||||||||||||
| Dichte |
0,98 g·cm−3 (20 °C)[2] | |||||||||||||||
| Schmelzpunkt | ||||||||||||||||
| Siedepunkt |
115 °C[2] | |||||||||||||||
| Dampfdruck | ||||||||||||||||
| pKS-Wert |
5,23 (konjugierte Säure bei 25 °C)[3] | |||||||||||||||
| Löslichkeit |
mischbar mit Wasser, Ethanol, Aceton, Chloroform, Diethylether und Benzol[4] | |||||||||||||||
| Dipolmoment | ||||||||||||||||
| Brechungsindex |
1,5095[4] | |||||||||||||||
| Sicherheitshinweise | ||||||||||||||||
| ||||||||||||||||
| MAK | ||||||||||||||||
| Toxikologische Daten | ||||||||||||||||
| Thermodynamische Eigenschaften | ||||||||||||||||
| ΔHf0 | ||||||||||||||||
| Soweit möglich und gebräuchlich, werden SI-Einheiten verwendet. Wenn nicht anders vermerkt, gelten die angegebenen Daten bei Standardbedingungen. Brechungsindex: Na-D-Linie, 20 °C | ||||||||||||||||


In der chemischen Industrie ist Pyridin sowohl ein bedeutender Synthesebaustein für die Arzneimittel- oder Herbizidherstellung als auch ein gängiges Lösungsmittel für chemische Reaktionen. Weltweit werden jährlich Zehntausende Tonnen der Verbindung hergestellt und zu großen Teilen in der chemischen Industrie weiterverwendet. Historisch wurde Pyridin aus Teer oder als Nebenprodukt der Kohlevergasung gewonnen; auf Grund des gestiegenen Bedarfs sind diese Methoden jedoch im Laufe der Jahre ökonomischeren synthetischen Verfahren gewichen.
Pyridin erfüllt die Hückel-Kriterien für Aromatizität und weist typische heteroaromatische Eigenschaften auf. Seine Reaktivität gegenüber elektrophilen Substitutionen ist gegenüber dem homoaromatischen Analogon Benzol deutlich herabgesetzt, wohingegen nukleophile Substitutionen häufiger auftreten.
Geschichte
.jpg.webp)
Pyridin wurde zweifellos bereits zu alchemistischen Zeiten durch Erhitzen tierischen Materials in unreiner Form erhalten.[10] Die früheste schriftliche Erwähnung im Jahr 1851 ist jedoch dem schottischen Naturwissenschaftler Thomas Anderson (1819–1874) zuzuschreiben.[11] Er untersuchte die Inhaltsstoffe von Knochenöl, das durch starkes Erhitzen trockener Knochen erhalten wird. Hierbei erhielt er unter anderem eine farblose, übelriechende Flüssigkeit, die er zwei Jahre später erstmals in reiner Form isolieren konnte.[12]
„Die erste dieser Basen, welche ich Pyridin nennen will, ist in der bei etwa 115 °C übergehenden Portion enthalten. Diese Portion besitzt einen dem des Picolins sehr ähnlichen, allein noch stärkeren und stechenderen Geruch. Sie ist vollkommen durchsichtig und farblos, und färbt sich in Berührung mit der Luft nicht. Sie ist in jedem Verhältniß in Wasser und leicht in flüchtigen und nicht flüchtigen Oelen löslich. In concentrirten Säuren löst sie sich unter starker Wärmeentwicklung, und bildet sehr leicht lösliche Salze mit denselben.“
Den Namen, der sich von griechisch πυρος (Pyros) = Feuer ableitet, erhielt Pyridin analog zu der bereits bekannten Stickstoffbase Pyrrol, da die erstmalige Isolierung ebenfalls bei hohen Temperaturen stattfand. Die Endung -in wurde im Einklang mit den bereits etablierten organischen Basen Anilin und Toluidin gewählt.
Die chemische Struktur von Pyridin konnte erst Jahrzehnte später endgültig aufgeklärt werden. Körner und Dewar postulierten unabhängig voneinander die Hypothese, dass eine Analogie zwischen Benzol und Naphthalin sowie Pyridin und Chinolin bestehe, in den Strukturen der erstgenannten müsse lediglich eine CH-Einheit durch ein Stickstoffatom ersetzt werden.[13] Dies konnte durch Reduktion von Pyridin mittels metallischen Natriums zu Piperidin, dessen Struktur zu dieser Zeit bereits bekannt war, bewiesen werden.
Im Jahre 1877 leitete William Ramsay Acetylen- und Blausäuregas durch ein rotglühendes Rohr, wobei Pyridin entstand.[14] Dies macht Pyridin zu einer der ersten synthetisch hergestellten heteroaromatischen Verbindungen.
In den folgenden Jahrzehnten wuchs der Bedarf an Pyridin, weshalb synthetische Methoden zu seiner Gewinnung entwickelt wurden. Ein Durchbruch gelang hierzu dem russischen Chemiker Alexei Jewgenjewitsch Tschitschibabin, der 1924 eine wirtschaftliche Syntheseroute aus kostengünstigen Synthesebausteinen entwickelte,[15] die auch heute noch zur industriellen Herstellung herangezogen wird.[8]
Vorkommen

Es sind nur wenige natürliche Vorkommen freien Pyridins bekannt. Es konnte jedoch in den flüchtigen Bestandteilen des Eibischs[16] sowie den Blättern und Wurzeln der Schwarzen Tollkirsche (Atropa belladonna)[17] nachgewiesen werden. Seine Derivate sind hingegen häufig Bestandteil von Biomolekülen wie den nach ihm benannten Pyridinnukleotiden und natürlichen Ölen und Gasen.
Pyridin entsteht durch Röst- und Konservierungsprozesse in Nahrungsmitteln und kann in geringen Mengen in deren flüchtigen Bestandteilen nachgewiesen werden. Hierzu gehören gebratenes Huhn,[18] Sukiyaki,[19] gebratener Schinken,[20] Beaufort-Käse,[21] Kaffeearoma,[22] schwarzer Tee[23] und Sonnenblumenhonig.[24] Sowohl der Rauch von Tabak[25][26] als auch von Marihuana[27] enthalten Pyridin.
Nomenklatur
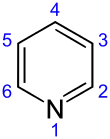
Die systematische Bezeichnung von Pyridin nach dem Hantzsch-Widman-System, das von der IUPAC empfohlen wird, ist Azin. Im Bereich der Heterocyclennomenklatur werden jedoch oftmals historisch gängige Trivialnamen benutzt, weshalb die systematische Bezeichnung weder im Sprachgebrauch noch in der Fachliteratur gängig ist. Entgegen der Systematik empfiehlt die IUPAC ausdrücklich die Beibehaltung der Bezeichnung Pyridin.[28] Die Nummerierung der Ringatome beginnt am Stickstoffatom, das die höchste Priorität besitzt, und setzt sich hiervon ausgehend von 2 bis 6 über die Kohlenstoffringglieder fort. Auch eine Zuweisung der Positionen durch Buchstaben des griechischen Alphabets (α–γ) und die Substitutionsmusternomenklatur, die in homoaromatischen Systemen üblich ist (ortho, meta, para) sind teilweise anzutreffen.
Die systematische Bezeichnung des Pyridinrestes lautet Pyridinyl, wobei die Position der Verknüpfung als Zahl vorangestellt wird. Pyridin stellt jedoch auch in diesem Fall eine Ausnahme der Systematik dar, da das historisch übliche Pyridyl als Bezeichnung empfohlen wird.[29] Der kationische Pyridinrest, der aus der Addition eines Elektrophils an das Stickstoffatom resultiert, wird Pyridinium bezeichnet.
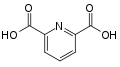 2,6-Pyridindicarbonsäure (Dipicolinsäure)
2,6-Pyridindicarbonsäure (Dipicolinsäure)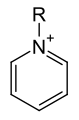 Grundstruktur von Pyridiniumverbindungen
Grundstruktur von Pyridiniumverbindungen
Gewinnung und Darstellung
Historisch wurde Pyridin aus Teer oder bei der Kohlevergasung gewonnen. Im Steinkohlenteer sind jedoch nur etwa 0,1 % Pyridin enthalten,[30] das als Gemisch mit weiteren Substanzen aus der Rohsubstanz ausgetrieben werden kann. Zur Auftrennung des Gemischs sind jedoch mehrstufige Reinigungsprozesse nötig, weshalb angesichts der geringen Ausbeute ein solches Verfahren nicht mehr wirtschaftlich ist. Heutzutage wird nahezu der gesamte weltweite Bedarf durch synthetisches Pyridin gedeckt.[8]
Tschitschibabin-Pyridinsynthese
Moderne industrielle Synthesen nutzen die von Tschitschibabin 1924 erstmals publizierte Route,[15] wobei es sich um eine Multikomponentenreaktion zwischen Ketonen oder Aldehyden mit Ammoniak handelt. Zur Synthese des unsubstituierten Pyridins werden Formaldehyd und Acetaldehyd benötigt – kostengünstige Synthesebausteine, die im Multitonnenmaßstab verfügbar sind. In einer Aldolkondensation wird hierbei aus je einem Teil der Aldehyde zunächst Acrolein gebildet, das mit Acetaldehyd und Ammoniak zu 1,4-Dihydropyridin kondensiert und dann am Festphasenkatalysator zu Pyridin oxidiert wird. Technisch wird dies als Gasphasenreaktion bei 400–450 °C durchgeführt. Die Zusammensetzung des Produktgemischs, bestehend aus Pyridin, einfach methylierten Pyridinen (Picoline) und Lutidinen, ist abhängig vom verwendeten Katalysator und kann den Bedürfnissen des Herstellers angepasst werden. Als Katalysatormaterialien dienen Übergangsmetallsalze wie Cadmiumfluorid und Mangan(II)-fluorid auf Silicatträgern, aber auch Cobalt- und Thallium-Verbindungen können zum Einsatz kommen. Das gewonnene Pyridin kann in einem mehrstufigen Prozess von den Nebenprodukten abgetrennt werden und diese können entweder weiterverarbeitet oder durch Demethylierung in Pyridin umgewandelt werden.[8]
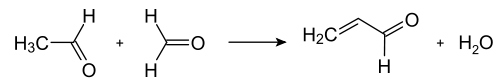
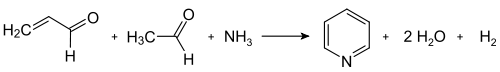
Dealkylierung von Alkylpyridinen
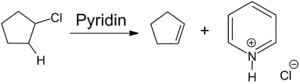
Pyridin kann durch Dealkylierung von alkylierten Pyridinen, die als Nebenprodukte in gängigen industriellen Synthesen anfallen, hergestellt werden. Die Dealkylierung verläuft entweder oxidativ mit Luft am Vanadiumoxid-Katalysator,[31] durch Dampfdealkylierung am Nickelkatalysator[32][33] oder durch Hydrodealkylierung am Silber- oder Platinkatalysator.[34] Hierbei sind Ausbeuten an Pyridin von bis zu 93 % am Nickelkatalysator möglich.[8]
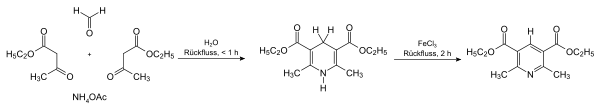
Hantzschsche Pyridinsynthese
Ein erster bedeutender Syntheseweg von Pyridinderivaten wurde 1881 von Arthur Hantzsch beschrieben.[35] Hierbei werden ein β-Ketoester (häufig Acetessigester), ein Aldehyd (häufig Formaldehyd) und Ammoniak beziehungsweise Ammoniumsalze im Verhältnis 2:1:1 eingesetzt (Hantzschsche Dihydropyridinsynthese). Es wird zunächst ein zweifach hydriertes Pyridin erhalten, das in einem anschließenden Schritt oxidativ zum entsprechenden Pyridinderivat umgesetzt werden kann. Knoevenagel zeigte, dass auch unsymmetrisch substituierte Pyridinderivate auf diesem Wege zugänglich sind.[36]
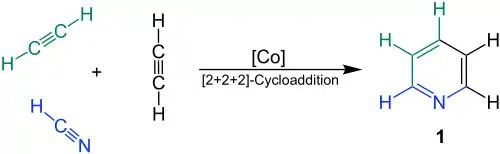
Bönnemann-Cyclisierung
Die Trimerisierung von einem Teil der Nitrilkomponente und zwei Teilen Acetylen wird nach Helmut Bönnemann als Bönnemann-Cyclisierung bezeichnet. Hierbei handelt es sich um eine Abwandlung der Reppe-Synthese, die sowohl thermisch als auch photochemisch durchgeführt werden kann. Während bei der thermischen Reaktion hohe Drücke und Temperaturen benötigt werden, kann die photoinduzierte Cycloaddition unter Normalbedingungen bei katalytischem Einsatz von CoCp2(cod) (Cp=Cyclopentadienyl, cod=1,5-Cyclooctadien) sogar in Wasser durchgeführt werden.[37] Auf diesem Wege sind eine Reihe von Pyridinderivaten zugänglich. Bei Verwendung von Acetonitril als Nitrilkomponente wird 2-Methylpyridin erhalten, das zu Pyridin dealkyliert werden kann.
Biosynthese des Pyridinrings
Mehrere Pyridinderivate treten in biologischen Systemen in teils prominenter Funktion auf. Der exakte biosynthetische Aufbau des Pyridinrings ist abhängig vom biologischen System und der genauen Struktur des Pyridinderivats. Während der biosynthetische Zugang vieler Pyridinderivate noch nicht vollständig geklärt ist, gilt der Syntheseweg des Pyridinderivats Nicotinsäure (Vitamin B3) in einigen Bakterien, Pilzen und Säugetieren als gesichert. Säugetiere synthetisieren Nicotinsäure oftmals durch oxidativen Abbau der Aminosäure Tryptophan, wobei als Zwischenprodukt das Anilinderivat Kynurenin entsteht. In den Bakterien Mycobacterium tuberculosis und Escherichia coli werden zur Biosynthese hingegen Glycerinaldehyd-3-phosphat und Asparaginsäure benötigt.[38]
Eigenschaften
Physikalische Eigenschaften
| Kritische Größen[39] | ||
|---|---|---|
| Druck | Temperatur | Volumen |
| 6,70 MPa | 620 K | 229 cm3·mol−1 |
| Parameter für die Antoine-Gleichung (340–426 °C)[40] | ||
| A | B | C |
| 4,16272 | 1371,358 | −58,496 |
| Temperaturabhängigkeit des Dampfdrucks[41] (nach ΔVH0=A·exp(−β·Tr)·(1−Tr)β) zwischen 298 und 388 °C | ||
| A | β | Tc |
| 55,43 kJ·mol−1 | 0,2536 | 620 K |
Pyridin ist farblos und bei Standardbedingungen flüssig, siedet bei 115,23 °C und gefriert bei −41,70 °C. Es ist eine stark lichtbrechende Flüssigkeit, die bei 20 °C und einer Wellenlänge von 589 nm einen Brechungsindex von 1,5095 aufweist. Bei Standardbedingungen besitzt Pyridin eine mit Wasser vergleichbare Dichte von 0,9819 g·cm−3.[4] Pyridin weist ein elektrisches Dipolmoment von 2,2 D auf[1], ist diamagnetisch und besitzt eine molare diamagnetische Suszeptibilität von −48,7·10−6 cm3·mol−1.[42] In der flüssigen Phase beträgt die Standardbildungsenthalpie 100,2 kJ·mol−1, in der Gasphase hingegen 140,4 kJ·mol−1.[9] Bei 25 °C besitzt Pyridin eine dynamische Viskosität[43] von 0,879 mPa·s[44] und eine Wärmeleitfähigkeit von 0,166 W·(m·K)−1.[45] Unter Standardbedingungen ergibt sich ein Dampfdruck von 20,5 hPa.[2] Die Verdampfungsenthalpie beträgt am Siedepunkt unter Normaldruck 35,09 kJ·mol−1.[41] Am Schmelzpunkt wird eine Schmelzenthalpie von 8,28 kJ·mol−1 realisiert.[46]
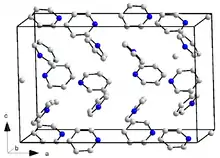
Pyridin kristallisiert im orthorhombischen Kristallsystem in der Raumgruppe Pna21 (Raumgruppen-Nr. 33) mit den Gitterparametern a = 1752 pm, b = 897 pm und c = 1135 pm und 16 Formeleinheiten pro Elementarzelle. Des Weiteren ist ein kristallines Trihydrat (Pyridin · 3 H2O) bekannt. Dieses kristallisiert ebenfalls im orthorhombischen Kristallsystem, jedoch in der Raumgruppe Pbca (Nr. 61) mit den Gitterparametern a = 1244 pm, b = 1783 pm und c = 679 pm und acht Formeleinheiten pro Elementarzelle.[47]
Chemische Eigenschaften
Pyridin ist mischbar mit Wasser, Ethanol, Diethylether, Aceton, Benzol und Chloroform.[4] Es reagiert schwach basisch und bildet mit Chlorwasserstoffsäure (Salzsäure) ein kristallines Hydrochlorid, das erst bei 145–147 °C schmilzt.[48]
Pyridin gehört zur Klasse der Heteroaromaten und weist typische Eigenschaften dieser Stoffklasse auf. Durch den Einfluss des elektronegativen Stickstoffs ist der Pyridinring jedoch relativ elektronenarm, wodurch die für aromatische Systeme typische elektrophile Substitutionsreaktion gehemmt wird. Im Vergleich zu seinem Kohlenstoffanalogon Benzol zeigt Pyridin somit eine deutlich geringere Reaktivität bezüglich elektrophiler aromatischer Substitutionen. Im Gegensatz zu Kohlenstoff-Aromaten besitzt Pyridin jedoch eine vergleichsweise höhere Reaktivität bezüglich nukleophiler Substitutionen und der Metallierung des Rings durch stark basische Organometallverbindungen.[49][50] Die Reaktivität von Pyridin weist Charakteristika dreier chemischer Gruppierungen auf. Mit Elektrophilen finden elektrophile Substitutionen statt, worin die aromatischen Eigenschaften von Pyridin zum Ausdruck kommen. Mit Nukleophilen reagiert Pyridin in 2- und 4-Position und weist somit Ähnlichkeiten mit der Reaktivität von Iminen oder Carbonylverbindungen auf. Die Reaktion mit vielen Lewis-Säuren führt zur Addition an das Stickstoffatom, wodurch Pyridin Ähnlichkeiten zur Reaktivität tertiärer Amine aufweist. Die Fähigkeit durch Oxidation N-Oxide zu bilden ist ebenfalls ein Charakteristikum tertiärer Amine.[51]
Mit zahlreichen Übergangsmetallionen bildet Pyridin Komplexe. Hierbei koordiniert Pyridin stark bevorzugt mit dem freien Elektronenpaar des Stickstoffatoms an das Metallzentrum. η6-Koordination, wie sie bei Benzol auftritt, ist nur durch sterische Blockierung des Stickstoffatoms möglich.[52]
Molekulare Eigenschaften
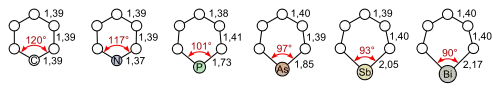
Pyridin weist ein durchkonjugiertes System von sechs π-Elektronen auf, die über das gesamte Ringsystem delokalisiert sind. Des Weiteren ist Pyridin planar gebaut und befolgt somit die Hückel-Kriterien für aromatische Systeme. Im Gegensatz zu Benzol ist die Elektronendichte jedoch nicht gleichmäßig verteilt, was auf den negativen induktiven Effekt des Stickstoffatoms zurückzuführen ist. Aus diesem Grund weist Pyridin ein Dipolmoment auf und ist schlechter resonanzstabilisiert als Benzol (Benzol: 150 kJ·mol−1, Pyridin: 117 kJ·mol−1).[53] Die höhere Elektronendichte drückt sich auch in der verkürzten Bindungslänge der Stickstoff-Kohlenstoff-Bindung aus (Benzol: 139 pm, Pyridin, C-N: 137 pm),[54] während die Kohlenstoff-Kohlenstoff-Bindungen die gleiche Bindungslänge wie im Benzolmolekül (139 pm) aufweisen. Die Bindungslängen verdeutlichen den aromatischen Charakter von Pyridin. Wie für aromatische Systeme üblich liegen sie zwischen den Werten, die typischerweise für einfachgebundene und doppeltgebundene Atome erwartet werden.
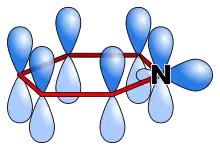
Im Pyridinmolekül sind alle Ringatome sp2-hybridisiert. Das Stickstoffatom stellt das Elektron seines p-Orbitals zur Ausbildung des aromatischen Systems zur Verfügung, sein freies sp2-Elektronenpaar liegt in der Molekülebene und weist vom Ring nach außen. Dieses Elektronenpaar trägt nicht zum aromatischen System bei, ist jedoch für die chemischen Eigenschaften von Pyridin von großer Bedeutung. Auf Grund der periplanaren Anordnung des freien Elektronenpaars wird das aromatische System durch Bindungsbildung an dieser Position nicht aufgehoben, was einen elektrophilen Angriff an dieser Position begünstigt. Die Trennung des freien Elektronenpaars von aromatischen System bewirkt jedoch auch, dass das Stickstoffatom keinen positiven mesomeren Effekt ausbilden kann. Die Reaktivität von Pyridin wird somit zu großen Teilen von dem negativen induktiven Effekt des Stickstoffatoms bestimmt.
Pyridin ist über fünf mesomere Grenzstrukturen resonanzstabilisiert und aus diesem Grund stabiler als das hypothetische 1-Aza-1,3,5-cyclohexatrien mit lokalisierten Doppelbindungen. Analog dem Benzol existieren zwei Grenzstrukturen, die keinen zwitterionischen Charakter besitzen. Zusätzlich können jedoch drei weitere zwitterionische Grenzstrukturen formuliert werden, die dem Stickstoffatom eine negative Ladung zuweisen, wodurch die positive Ladung an der 4-Position beziehungsweise einer der beiden 2-Positionen des Rings auftritt. Die Lage der Ladung am Stickstoffatom steht im Einklang mit dessen höherer Elektronegativität im Vergleich zu Kohlenstoff.
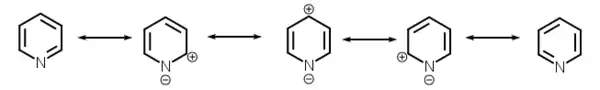 Mesomere Grenzstrukturen von Pyridin.
Mesomere Grenzstrukturen von Pyridin.
Reaktionen
Viele für das homologe Benzol charakteristische Reaktionen laufen an Pyridin nicht oder nur unter aufwändigeren Bedingungen beziehungsweise mit schlechter Ausbeute ab. Hierfür sind im Wesentlichen die verringerte Elektronendichte im aromatischen System, die Pyridin und dessen Derivate für elektrophile Substitutionen desaktiviert, sowie die bevorzugte Addition von Elektrophilen am elektronenreichen Stickstoffatom verantwortlich. Die elektrophile Addition am Stickstoffatom führt zu einer weiteren Desaktivierung des Aromaten, wodurch anschließende elektrophile Substitutionen nochmals erschwert sind. Auf der anderen Seite treten radikalische und nukleophile Substitutionen im Vergleich zu Benzol häufiger auf und stellen oft sogar den bevorzugten Reaktionsweg dar.[1][55]
Elektrophile Substitutionen
Elektrophile Substitutionen an Pyridin laufen in vielen Fällen nicht oder nur unvollständig ab, der Heteroaromat kann jedoch durch elektronenschiebende Funktionalisierung aktiviert werden. Gängige Alkylierungen und Acylierungen (beispielsweise durch Friedel-Crafts-Alkylierung oder -Acylierung) versagen meist, da sie nur zur Addition am Stickstoffatom führen. Substitutionen finden in der Regel an der 3-Position statt, da es sich zum einen um das elektronenreichste Kohlenstoffatom des Moleküls handelt, wodurch eine elektrophile Addition erleichtert wird, zum anderen besitzt der entstehende σ-Komplex keine Grenzstruktur, in der beim Stickstoffatom die Oktettregel verletzt wird. Dies ist im Falle einer Addition in 2- oder 4-Position der Fall und bewirkt somit einen energetisch ungünstigeren σ-Komplex.
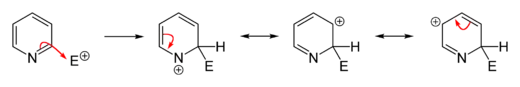
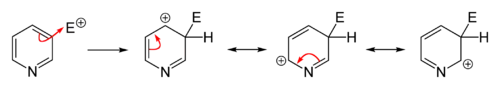

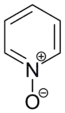
Sollen jedoch Substituenten in 2- oder 4-Position eingeführt werden, so existieren etablierte Möglichkeiten, die Reaktion entsprechend zu führen. Eine häufig angewandte Variante ist die Durchführung der elektrophilen Substitution am aktivierten N-oxid des Pyridins und anschließender Desoxygenierung des Stickstoffatoms. Bei dieser Variante werden in der Regel in 2- und 4-Position substituierte Produkte erhalten, da das Sauerstoffatom des N-oxids dem aromatischen System Elektronendichte zur Verfügung stellt und hiermit die Substitution an diesen Positionen gegenüber einer Substitution in 3-Position begünstigt. Zur Desoxygenierung können eine Reihe gängiger Reduktionsmittel verwendet werden. Es eignen sich allgemein dreiwertige Phosphorverbindungen oder zweiwertige Schwefelverbindungen, die leicht oxidierbar sind. Als günstiges Reagenz wird häufig Triphenylphosphan eingesetzt, das in der Reaktion zu Triphenylphosphinoxid oxidiert wird. Im Folgenden werden ausgewählte verbreitete elektrophile Substitutionen an Pyridin exemplarisch erläutert.[55]
Die direkte Nitrierung von Pyridin läuft selbst unter drastischen Bedingungen nur mit sehr geringen Ausbeuten ab. 3-Nitropyridin kann jedoch auf anderem Wege durch Reaktion von Pyridin mit Distickstoffpentoxid und Natriumhydrogensulfit hergestellt werden.[56][57] Pyridinderivate, die das Stickstoffatom sterisch und/oder elektronisch abschirmen, können durch Nitroniumtetrafluoroborat direkt nitriert werden. Auf diesem Wege gelingt die Synthese von 3-Nitropyridin aus 2,6-Dibrompyridin und anschließender Dehalogenierung.[58] Mit ähnlichem Erfolg verläuft auch die Sulfonierung von Pyridin, die selbst unter scharfen Bedingungen ohne nennenswerten Umsatz abläuft. Pyridin-3-sulfonsäure entsteht jedoch durch Kochen in einem Überschuss von Oleum bei 320 °C mit akzeptabler Ausbeute.[59] Die Begründung für dieses Verhalten ist die bevorzugte Addition des Elektrophils Schwefeltrioxid an den Pyridinstickstoff, wodurch der Heteroaromat für den zur Einführung der Sulfonsäuregruppe benötigten elektrophilen Angriff zusätzlich desaktiviert wird.[55] Die Sulfonierung mit Oleum verläuft jedoch glatt in Gegenwart katalytischer Mengen von Quecksilber(II)-sulfat.[60] Der zu Grunde liegende Mechanismus ist bisher nicht geklärt.[55]
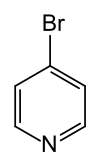
Im Gegensatz zur Nitrierung und Sulfonierung sind die Bromierung und Chlorierung von Pyridin auf direktem Wege möglich. Die Umsetzung von Pyridin mit molekularem Brom in Oleum bei 130 °C zu 3-Brompyridin verläuft mit sehr guter, die Chlorierung mit molekularem Chlor in Gegenwart von Aluminiumchlorid bei 100 °C zu 3-Chlorpyridin hingegen nur mit mäßiger Ausbeute.[55] In Gegenwart katalytischer Mengen an Palladium(II)-chlorid sind auch 2-Brompyridin sowie 2-Chlorpyridin durch Reaktion mit den molekularen Halogenen präparativ zugänglich.[55]
Nukleophile Substitutionen
Im Gegensatz zu Benzol sind eine Reihe effizienter nukleophiler Substitutionen an Pyridin bekannt. Der Grund hierfür ist die vergleichsweise geringere Elektronendichte des Heteroaromaten, der Angriffe durch Nukleophile begünstigt. Hierbei treten sowohl ipso-Substitutionen an Abgangsgruppen-tragenden Ringatomen als auch Reaktionen unter Abspaltung von Hydridionen sowie Eliminierungs-Additions-Reaktionen über Heteroarin Zwischenstufen auf. Sie liefern meist die in 2- oder 4-Position substituierten Produkte.[50][55]
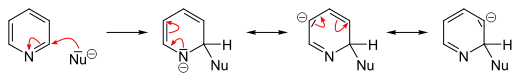
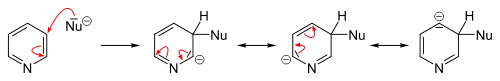

An Pyridinderivaten, die gute Abgangsgruppen tragen, laufen ipso-Substitutionen in vielen Fällen glatt ab. Hierzu dienen meist brom-, chlor- oder fluorsubstituierte Substrate, aber auch die Sulfonsäuregruppe kann als Abgangsgruppe dienen. Für die Substitution mit Organolithium-Verbindungen ist Fluor die beste Abgangsgruppe. Als Nukleophile können des Weiteren Alkoholate, Thiolate aber auch Amine, bei erhöhtem Druck auch Ammoniak, eingesetzt werden.[49]
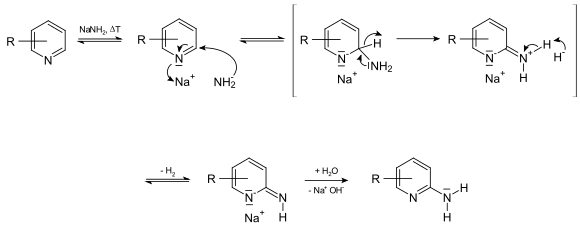
Das Hydridion ist allgemein eine sehr schlechte Abgangsgruppe. In der Heterocyclenchemie sind jedoch wenige Reaktionen bekannt, bei denen das Hydridion als Abgangsgruppe fungiert. Hierzu zählt die Tschitschibabin-Reaktion, durch die in 2-Position aminierte Pyridinderivate hergestellt werden können. Als Nukleophil wird hierzu Natriumamid eingesetzt das in 2-Position an Pyridin addiert und nach wässriger Aufarbeitung der Reaktion 2-Aminopyridin freisetzt. Das Hydridion wird im Verlauf der Reaktion vom Pyridinring abgespalten und bildet mit einem Proton einer weiteren Aminogruppe molekularen Wasserstoff.[50][61]
Bei der Verwendung von Lithiumorganylen als Nukleophile können diese direkt, auf Grund der dirigierenden Wirkung des Stickstoffatoms bevorzugt in 2-Position, an Pyridin addieren. Je nach verwendetem Nukleophil und Substrat kommt es anschließend zur direkten Abspaltung von Lithiumhydrid. Ist das intermediär entstehende N-Lithiumsalz jedoch persistent, müssen zur Rearomatisierung unter Freisetzung des substituierten Pyridins oxidative Bedingungen geschaffen werden.[49][50]
Analog zu Benzol ist die Bildung von Heteroarinen als Zwischenprodukt möglich. Hierzu werden Pyridinderivate mit guten Abgangsgruppen mit starken Basen wie Natriumamid und Kalium-tert-butanolat zum Heteroarin eliminiert. Die anschließende Addition eines Nukleophils an der Dreifachbindung verläuft meist mit geringer Selektivität und es wird ein Gemisch aus den beiden möglichen Additionsprodukten erhalten.[49]
Radikalische Reaktionen
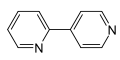
An Pyridin laufen verschiedene radikalische Reaktionen ab. Von präparativem Interesse sind hierbei Dimerisierungen von Pyridin zu Bipyridinen. Die radikalische Dimerisierung von Pyridin mit elementarem Natrium beziehungsweise Raney-Nickel liefert selektiv 4,4′-Bipyridin[62] beziehungsweise 2,2′-Bipyridin,[63] die bedeutende Grundstoffe in der chemischen Industrie darstellen. Als Namensreaktionen sind radikalische Reaktionen unter sauren Bedingungen an Heteroaromaten als Minisci-Reaktion bekannt. An Pyridin führen diese mit hoher Selektivität zu den in 2- beziehungsweise 4-Position substituierten Produkten. So ist 2-tert-Butylpyridin aus Pyridin durch Reaktion mit Pivalinsäure, Silbernitrat und Ammoniumperoxodisulfat in schwefelsaurer Lösung mit einer Ausbeute von 97 % durch eine Minisci-Reaktion zugänglich.[49]
Reaktionen am Stickstoffatom
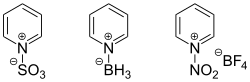
Lewis-Säuren addieren leicht an das Stickstoffatom von Pyridin, wobei Pyridiniumsalze gebildet werden. Mit Halogenwasserstoffsäuren werden analog die entsprechenden Hydrochloride oder Hydrobromide erhalten, denen größere Bedeutung zukommt. Die Reaktion mit Alkylhalogeniden führt zur Alkylierung des Stickstoffatoms. Hierdurch entsteht eine positive Ladung im Ring, welche die Reaktivität von Pyridin stark beeinflusst und sowohl Oxidations- als auch Reduktionsreaktionen erleichtert. Zur selektiven Einführung von Resten an Pyridiniumverbindungen kann die Zincke-Reaktion verwendet werden, wobei die zu Grunde liegenden primären Amine benötigt werden.
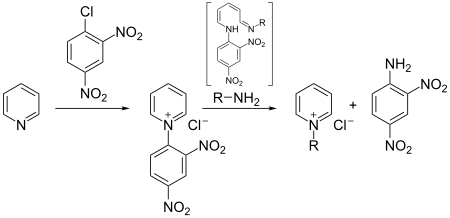
Die Reaktion mit sekundären Aminen führt hingegen zur Ringöffnung, wobei Zincke-Aldehyde erhalten werden.
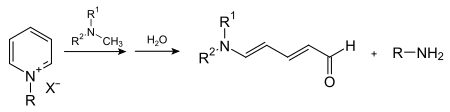
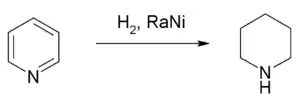
Hydrierung und Reduktion
Durch vollständige Hydrierung mittels Wasserstoff in Gegenwart von Raney-Nickel wird das gesättigte Piperidin erhalten.[64] Dabei wird eine Reaktionswärme von −193,8 kJ·mol−1 freigesetzt.[65] Diese ist etwas geringer als die Hydrierwärme von Benzol mit −205,3 kJ·mol−1.[65]
Unter milderen Bedingungen können teilhydrierte Derivate erhalten werden. So ergibt die Reduktion mittels Lithiumaluminiumhydrid ein Gemisch aus 1,4-Dihydropyridin, 1,2-Dihydropyridin und 2,5-Dihydropyridin.[66] Reines 1,4-Dihydropyridin bildet sich aus Pyridin in Gegenwart organischer Magnesium- und Zink-Komplexe.[67] (Δ3,4)-Tetrahydropyridin ist durch elektrochemische Reduktion von Pyridin zugänglich.[68]
Verwendung

Heute ist Pyridin ein bedeutender Grundstoff in der chemischen Industrie, der jährlich im Kilotonnenmaßstab hergestellt wird (26.000 t/a, Stand 1989).[8] Es sind weltweit 25 Produktionsstandorte für Pyridin bekannt, von denen sich elf auf europäischem Boden befinden (Stand: 1999).[27] Zu den bedeutenden Produzenten von Pyridin zählen oder zählten Degussa, Rütgerswerke, ICI und Koei Chemical.[8] In den vergangenen Jahren wurde die Kapazität zur Pyridinproduktion jedoch erheblich gesteigert, sodass allein in der Volksrepublik China Anlagen mit einer Kapazität von 50.000 t/a entstanden sind.[69] Nach eigenen Angaben ist das amerikanisch-chinesische Joint-Venture Vertellus derzeit Weltmarktführer für Pyridin.[70]
Pyridin besitzt weite Anwendungsgebiete in der präparativen chemischen Industrie. Es wird als polares, basisches, wenig reaktives Lösungsmittel verwendet,[27] das sowohl als Katalysator, aktivierendes Agens als auch als Base zum Abbinden entstehender Säuren verwendet wird. Es eignet sich insbesondere zur Dehalogenierung, wobei es als Base der Eliminierungsreaktion fungiert und die entstehende Halogenwasserstoffsäure unter Bildung eines Pyridiniumsalzes abbindet. In Veresterungen und Acylierungen kann Pyridin zur Aktivierung der eingesetzten Carbonsäurehalogenide oder -anhydride eingesetzt werden. Eine höhere Aktivität bei diesen Reaktionen weisen jedoch die Pyridinderivate DMAP und PPY auf. Auch in Kondensationsreaktionen kann Pyridin als Base eingesetzt werden.
Das Chromatsalz Pyridiniumchlorochromat (PCC) wurde 1975 von Elias Corey und William Suggs entwickelt und dient als starkes Oxidationsmittel, das meist zur Oxidation von Alkoholen eingesetzt wird.[71] Es wird aus der Reaktion von Pyridin mit Salzsäure und Chrom(VI)-oxid erhalten. Da es krebserregend ist, sollte es jedoch möglichst durch weniger toxische Oxidationsmittel ersetzt werden. Das Cornforth- (Pyridiniumdichromat, PDC) und das Collins-Reagenz sind ähnliche Chrom-basierte Pyridinverbindungen, die das gleiche Gefahrenpotential bergen und ebenfalls zur Oxidation eingesetzt werden.
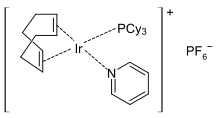
In Metallkomplexen ist Pyridin ein labiler Ligand und kann leicht durch stärker komplexierende Lewis-Basen ausgetauscht werden, was in der Katalyse ausgenutzt wird. Pyridinkomplexe mit Übergangsmetallionen finden als Polymerisations-[72][73] oder Hydrierkatalysatoren, beispielsweise dem Crabtree-Katalysator,[74] Verwendung. Die Katalysatorspezies trägt zunächst einen Pyridinliganden, der leicht durch das Substrat ausgetauscht wird. Nach Beendigung des Katalysezyklus koordiniert Pyridin wieder am Katalysator und bewirkt so die koordinative Absättigung des Metallions.
In der chemischen und pharmazeutischen Industrie dient Pyridin als Synthesebaustein zur Herstellung einer Vielzahl von Arzneistoffen, Insektiziden und Herbiziden. In großen Mengen wird oder wurde Pyridin zur Herstellung der Herbizide Diquat oder Paraquat verwendet, die ein Bipyridingerüst besitzen. Der erste Syntheseschritt zum Insektizid Chlorpyrifos besteht aus der Chlorierung von Pyridin, ebenso ist es die Ausgangsverbindung zur Herstellung des Fungizids Pyrithion.[27] Die quartären Pyridiniumsalze Cetylpyridiniumchlorid und Laurylpyridiniumchlorid, die in einer Zincke-Reaktion aus Pyridin hergestellt werden können, werden als Antiseptikum in Mund- und Zahnpflegemitteln eingesetzt.[1]
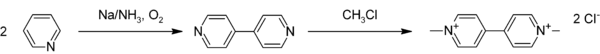
Neben Pyridinen sind auch Derivate des Piperidins wichtige Synthesebausteine. Eine gängige Synthese von Piperidin besteht in der Reduktion von Pyridin. In industriellen Verfahren kann Pyridin am Nickel-, Cobalt- oder Rutheniumkatalysator bei erhöhter Temperatur glatt zu Piperidin reduziert werden.[76]
Unter anderem wird Pyridin auch in der Farbstoff- und Gummiproduktion als Lösungsmittel eingesetzt[77] und in der Textilindustrie zur Verbesserung der Netzfähigkeit von Baumwolle verwendet.[1]
Zur Vergällung von Ethanol zu Brennspiritus werden dem Alkohol Substanzen beigemengt, die diesen für den Menschen ungenießbar werden lassen und nur schwer durch physikalische Verfahren abzutrennen sind. Pyridin war auf Grund seines bitteren Geschmacks und seiner physikalischen Eigenschaften häufig Bestandteil dieses Stoffgemischs, ist heutzutage jedoch meist durch andere Stoffe ersetzt.[1] In geringer Dosierung wird Pyridin jedoch auch als bitterer Geschmacksstoff in Nahrungsmitteln verwendet. In Lösung liegt der Erkennungsschwellwert von Pyridin bei 1–3 mmol·l−1 (79–237 mg·l−1).[78]
In seiner Eigenschaft als Base kann Pyridin als Bestandteil des Karl-Fischer-Reagenz eingesetzt werden. In modernen Reagenzien ist es jedoch meist auf Grund der Geruchsbelästigung durch eine andere Base ausgetauscht.[79]
Gefahrenhinweise
Pyridin weist einen Flammpunkt von 17 °C auf und ist folglich leichtentzündlich. Die Zündtemperatur ist mit 550 °C angegeben. In einem Bereich von 1,7–10,6 Vol-% bildet Pyridin mit Luft explosive Gemische. Die thermische Zersetzung von Pyridin beginnt oberhalb von 490 °C, wobei als Zersetzungsprodukte Bipyridine, im Wesentlichen 2,2′-Bipyridin und in untergeordnetem Maße 2,3′-Bipyridin und 2,4′-Bipyridin, sowie Stickoxide und Kohlenstoffmonoxid gebildet werden.[2] Pyridin ist ferner als gesundheitsschädlich und wassergefährdend Klasse 2 eingestuft.[2] In aquatischen Systemen schädigt Pyridin sowohl tierische als auch pflanzliche Organismen und ist auf Grund seiner Mischbarkeit mit Wasser gut verfügbar.[80] Die erlaubte Maximale Arbeitsplatz-Konzentration (MAK) in den DACH-Staaten beträgt 5 ppm.[81]
Toxikologie
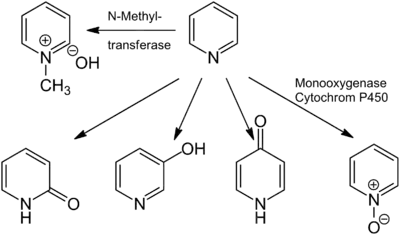
Der Kontakt mit Pyridin reizt Schleimhäute und die Haut und es treten Befindlichkeitsstörungen vor allem bezüglich des Magen-Darm-Traktes auf. Ferner weist Pyridin eine geringe neurotoxische Wirkung auf. Eine chronische Exposition mit Pyridin kann außerdem Störungen der Leber- und Nierenfunktion hervorrufen. In mehreren Versuchsreihen konnten die Genotoxizität und Clastogenität von Pyridin ausgeschlossen werden.[2][27][82] Die IARC stufte Pyridin im Jahr 2017 als möglicherweise krebserzeugend ein.[83]
In den meisten Fällen erfolgt die Aufnahme von Pyridin inhalativ, was zur Resorption in der Lunge führt. Die perorale Aufnahme führt hingegen zur Resorption im Gastrointestinaltrakt. Pyridin wird entweder unverändert oder metabolisiert über Kot oder Urin ausgeschieden. Durch Metabolisierung treten als Hauptprodukte N-Methylpyryliumhydroxid, das durch N-Methyltransferasen gebildet wird, sowie die Oxidationsprodukte Pyridin-N-oxid und 2-, 3- und 4-Hydroxypyridin, die durch Einwirkung von Monooxygenasen entstehen, auf. Der Mensch metabolisiert Pyridin jedoch ausschließlich zu N-Methylpyryliumhydroxid.[2][82]
Die Aufnahme toxischer Dosen von Pyridin verursacht Schwächegefühle, Ataxie, Salivation und kann Bewusstlosigkeit hervorrufen. Aus dem Jahr 1893 ist ein Todesfall nach versehentlicher oraler Aufnahme einer halben Tasse Pyridin bekannt.[27] Die niedrigste bekannte Letale Dosis (LDLo) für die orale Aufnahme von Pyridin bei Menschen beträgt 500 mg·kg−1. Pyridin hat in höheren Konzentrationen narkotisierende Wirkung und stellt ab einer Dampfkonzentration von 3600 ppm ein ernsthaftes Gesundheitsrisiko dar.[8]
Pyridin in der Umwelt
In geringen Mengen wird Pyridin bei industriellen Prozessen freigesetzt und an die Umwelt abgegeben. Es fällt in Spuren bei der Stahlerzeugung,[84] Kohlevergasung, in Kokereien, der Müllverbrennung und bei Verarbeitung von Ölschiefer an.[27] In der Umgebungsluft eines Ölschiefer-verarbeitenden Betriebes wurden Pyridinkonzentrationen von bis zu 13 µg·m−3 [85] beziehungsweise 53 µg·m−3 im Grundwasser in der Umgebung einer Kohlevergasungsanlage festgestellt.[86] Nach einer Untersuchung stehen 43.000 US-amerikanische Arbeiter potentiell im Kontakt mit Pyridin.[87]
Nachweis
Das UV/Vis-Spektrum von Pyridin in Hexan weist drei Absorptionsbanden auf. Diese korrespondieren mit einem π→π*-Übergang bei einer Wellenlänge von 195 nm (Extinktionskoeffizient ε = 7500 l·(mol·cm)−1), einem weiteren π→π*-Übergang bei 251 nm (ε = 2000 l·(mol·cm)−1) und einem n→π*-Übergang bei 270 nm (ε = 450 l·(mol·cm)−1).[55]
Im 1H-NMR-Spektrum von Pyridin weisen die Protonen ausgeprägte Tieffeldverschiebungen auf. Das Spektrum zeigt drei Signale korrespondierend mit den drei chemisch verschiedenen Protonen im Molekül. Die Signalintegrale stehen im Verhältnis 2:1:2. Das Signal bei tiefstem Feld resultiert von den α-Protonen δ(α-H) = 8,5 ppm, gefolgt von dem γ-Proton δ(γ-H) = 7,5 ppm und den β-Protonen δ(β-H) = 7,1 ppm. Benzol als carbocyclisches Analogon weist ein Protonensignal bei δ = 7,27 ppm auf. Die größeren chemischen Verschiebungen der α- und γ-Protonen im Vergleich zu Benzol resultieren aus der geringeren Elektronendichte im Pyridinring und korrespondieren relativ mit den niedrigeren Elektronendichten in α- und γ-Position, die aus den mesomeren Grenzstrukturen abgeleitet werden können. Die chemischen Verschiebungen der 13C-Kerne verhalten sich analog den Protonensignalen (δ(α-C) = 150 ppm, δ(β-C) = 124 ppm, δ(γ-C) = 136 ppm). Das 13C-Signal des Benzols liegt hingegen bei 129 ppm. Alle Werte beziehen sich auf lösungsmittelfreie Substanzen.[55]
Zur quantitativen Bestimmung der Pyridinkonzentration in der Umweltanalytik werden in der Regel gaschromatographische oder gekoppelte gas- und massenspektrometrische Methoden angewandt.[27]
Literatur
- E. Klingsberger: Pyridine - and Its Derivatives, 1. Auflage, Interscience Publishers, New York, 1960, ISBN 0-470-37917-0.
- T. Eicher, S. Hauptmann: The Chemistry of Heterocycles, 2. Auflage, Wiley-VCH, Weinheim, 2003, ISBN 3-527-30720-6.
- J. A. Joule, K. Mills: Heterocyclic Chemistry, 3. Auflage, Blackwell Science, Oxford, 2004, ISBN 0-632-05453-0.
- D. T. Davies: Basistexte Chemie: Aromatische Heterocyclen, 1. Auflage, Wiley-VCH, Weinheim 1995, ISBN 3-527-29289-6.
Weblinks
Einzelnachweise
- Eintrag zu Pyridin. In: Römpp Online. Georg Thieme Verlag, abgerufen am 27. Juli 2017.
- Eintrag zu Pyridin in der GESTIS-Stoffdatenbank des IFA, abgerufen am 20. Januar 2022. (JavaScript erforderlich)
- David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press/Taylor and Francis, Boca Raton, FL, Analytical Chemistry, S. 8-44.
- David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press/Taylor and Francis, Boca Raton, FL, Physical Constants of Organic Compounds, S. 3-448.
- Eintrag zu Pyridine im Classification and Labelling Inventory der Europäischen Chemikalienagentur (ECHA), abgerufen am 1. Februar 2016. Hersteller bzw. Inverkehrbringer können die harmonisierte Einstufung und Kennzeichnung erweitern.
- MAK Dokumentation für Pyridin, 2009, doi:10.1002/3527600418.mb11086d0047 (freier Volltext)
- Schweizerische Unfallversicherungsanstalt (Suva): Grenzwerte – Aktuelle MAK- und BAT-Werte (Suche nach 110-86-1 bzw. Pyridin), abgerufen am 2. November 2015.
- S. Shimizu, N. Watanabe, T. Kataoka, T. Shoji, N. Abe, S. Morishita, H. Ichimura: Pyridine and Pyridine Derivatives, in: Ullmann's Encyclopedia of Industrial Chemistry, 2005, Wiley-VCH Weinheim.
- David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press/Taylor and Francis, Boca Raton, FL, Thermochemistry, Electrochemistry, and Solution Chemistry, S. 5-28.
- A. Weissberger (Hrsg.), A. Klingsberg (Hrsg.), R. A. Barnes, F. Brody, P. R. Ruby: Pyridine and its Derivatives, Bd. 1, 1960, Interscience Pub. Inc. New York.
- Th. v. Anderson: Producte der trocknen Destillation thierischer Materien, in: Liebigs Ann., 1849, 70, S. 32–38; doi:10.1002/jlac.18490700105.
- Th. Anderson: Ueber die Producte der trocknen Destillation thierischer Materien, in: Liebigs Ann., 1851, 80, S. 44–65; doi:10.1002/jlac.18510800104.
- A. Ladenburg: Lectures on the history of the development of chemistry since the time of Lavoisier. Englische Übersetzung einer Vorlesung. Volltextzugriff (PDF; 5,0 MB).
- Über W. Ramsays Entdeckung in: Ber. Dtsch. Chem. Ges., 1877, 10, S. 736; doi:10.1002/cber.187701001202.
- A. Tschitschibabin in: J. Prakt. Chem., 1924, 107, S. 122.
- A. Täufel, W. Ternes, L. Tunger, M. Zobel: Lebensmittel-Lexikon, 4. Auflage, S. 450, Behr Verlag, 2005, ISBN 3-89947-165-2.
- G. A. Burdock (Hrsg.): Fenaroli’s Handbook of Flavor Ingredients, Band II, 3. Auflage, CRC Press, Boca Raton, 1995, ISBN 0-8493-2710-5.
- J. Tang, Q. Z. Jin, G.-H. Shen, C.-T. Ho, S. S. Cheng: Isolation and identification of volatile compounds from fried chicken, in: J. Agric. Food Chem., 1983, 31, S. 1287–1292; doi:10.1021/jf00120a035.
- T. Shibamoto, Y. Kamiya, S. Mihara: Isolation and identification of volatile compounds in cooked meat: sukiyaki, in: J. Agric. Food Chem., 1981, 29, S. 57–63; doi:10.1021/jf00103a015.
- C.-T. Ho, K. N. Lee, Q. Z. Jin: Isolation and identification of volatile flavor compounds in fried bacon, in: J. Agric. Food Chem., 1983, 31, S. 336–342; doi:10.1021/jf00116a038.
- J.-P. Dumont, J. Adda: Occurrence of sesquiterpenes in mountain cheese volatiles, in: J. Agric. Food Chem., 1978, 26, S. 364–367; doi:10.1021/jf60216a037.
- H. U. Aeschbacher, U. Wolleb, J. Löliger, J. C. Spadone, R. Liardon: Contribution of coffee aroma constituents to the mutagenicity of coffee, in: Food Chem. Toxicol., 1989, 27, S. 227–231.
- O. Vitzthum, P. Werkhoff, P. Hubert: New volatile constituents of black tea aroma, in: J. Agric. Food Chem., 1975, 23, S. 999–1003; doi:10.1021/jf60201a032.
- A. Täufel, W. Ternes, L. Tunger, M. Zobel: Lebensmittel-Lexikon, 4. Auflage, S. 226, Behr Verlag, 2005, ISBN 3-89947-165-2.
- M. Curvall, C. R. Enzell, B. Pettersson: An evaluation of the utility of four in vitro short term tests for predicting the cytotoxicity of individual compounds derived from tobacco smoke, in: Cell Biol. Toxicol., 1984, 1, S. 173–193; doi:10.1007/BF00125573.
- I. Schmeltz, D. Hoffmann: Nitrogen-containing compounds in tobacco and tobacco smoke, in: Chem. Rev., 1977, 77, S. 295–311; doi:10.1021/cr60307a001.
- Studie der Occupational Safety & Health Administration, OSHA, Washington, D.C., 1985. (PDF; 95 kB)
- W. H. Powell: Revision of the extended Hantzsch-Widman System of nomenclature for heteromonocycles. in: Pure Appl. Chem., 1983, 55, S. 409–416, Artikel (pdf; 187 kB).
- D. Hellwinkel: Die systematische Nomenklatur der Organischen Chemie, 4. Auflage, S. 45, Springer Verlag, Berlin, 1998, ISBN 3-540-63221-2.
- A. Gossauer: Struktur und Reaktivität der Biomoleküle, 2006, S. 488, Wiley-VCH Weinheim, ISBN 3-906390-29-2.
- ICI DE-AS 1917037, 1968.
- Nippon Kayaku, JP 7039545, 1967.
- Koei Chemicals, BE 758201, 1969.
- F. Mensch in: Erdöl Kohle Erdgas Petrochemie, 1969, 2, S. 67–71.
- A. Hantzsch: Condensationprodukte aus Aldehydammoniak und Ketonartigen Verbindungen, in: Chem. Ber., 1881, 14, S. 1637–1638, doi:10.1002/cber.18810140214.
- E. Knoevenagel, A. Fries: Synthesen in der Pyridinreihe. Ueber eine Erweiterung der Hantzsch'schen Dihydropyridinsynthese, in: Chem. Ber., 1898, S. 761–767, doi:10.1002/cber.189803101157.
- A. Behr: Angewandte homogene Katalyse, 2008, S. 722, Wiley-VCH Weinheim, ISBN 3-527-31666-3.
- J. B. Tarr, J. Arditti: Niacin Biosynthesis in Seedlings of Zea mays, in: Plant Physiol., 1982, 69, S. 553–556; doi:10.1104/pp.69.3.553.
- David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press/Taylor and Francis, Boca Raton, FL, Fluid Properties, S. 6-67.
- J. P. McCullough, D. R. Douslin, J. F. Messerly, I. A. Hossenlopp, T. C. Kincheloe, G. Waddington: Pyridine: experimental and calculated chemical thermodynamic properties between 0 and 1500 K., a revised vibrational assignment, in: J. Am. Chem. Soc., 1957, 79, S. 4289–4295; doi:10.1021/ja01573a014.
- V. Majer, V. Svoboda: Enthalpies of Vaporization of Organic Compounds: A Critical Review and Data Compilation, Blackwell Scientific Publications, Oxford, 1985, ISBN 0-632-01529-2.
- David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press/Taylor and Francis, Boca Raton, FL, Physical Constants of Organic Compounds, S. 3-673.
- David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press/Taylor and Francis, Boca Raton, FL, Fluid Properties, S. 6-211.
- Vertellus: Sicherheitsdatenblatt Pyridin. (PDF) In: https://www.vertellus.com/. Vertellus, 1. März 2018, abgerufen am 27. Mai 2021.
- David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press/Taylor and Francis, Boca Raton, FL, Fluid Properties, S. 6-221.
- E. S. Domalski, E. D. Hearing: Heat Capacities and Entropies of Organic Compounds in the Condensed Phase. Volume III, in: J. Phys. Chem. Ref. Data, 1996, 25, S. 1–525; doi:10.1063/1.555985.
- D. Mootz, H.-G. Wussow: Crystal structures of pyridine and pyridine trihydrate, in: J. Chem. Phys., 1981, 75, S. 1517–1522; doi:10.1063/1.442204.
- Datenblatt Pyridin bei AlfaAesar, abgerufen am 26. Juni 2010 (PDF) (JavaScript erforderlich).
- J. A. Joules, K. Mills: Heterocyclic Chemistry, 5. Auflage, S. 125–141, Blackwell Publishing, Chichester, 2010, ISBN 978-1-4051-9365-8.
- D. T. Davies: Basistexte Chemie: Aromatische Heterocyclen, 1. Auflage, Wiley-VCH, Weinheim 1995, ISBN 3-527-29289-6.
- R. Milcent, F. Chau: Chimie organique hétérocyclique: Structures fondamentales, 1. Auflage, S. 241–282, EDP Sciences, 2002, ISBN 2-86883-583-X.
- C. Elschenbroich: Organometallchemie, 6. Auflage, S. 524–525, Vieweg+Teubner Verlag, 2008, ISBN 3-8351-0167-6.
- J. A. Joule, K. Mills: Heterocyclic Chemistry, 5. Auflage, S. 7, Blackwell Publishing, Chichester, 2010, ISBN 1-4051-3300-7.
- C. Elschenbroich: Organometallchemie, 6. Auflage, S. 218, Vieweg+Teubner Verlag, 2008, ISBN 3-8351-0167-6.
- J. A. Joule, K. Mills: Heterocyclic Chemistry, 3. Auflage, 2004, Blackwell Science, Oxford, ISBN 0-632-05453-0.
- J. M. Bakke, I. Hegbom: Dinitrogen Pentoxide–Sulfur Dioxide, a New Nitration System, in: Acta Chemica Scandinavica, 1994, 48, S. 181–182; doi:10.3891/acta.chem.scand.48-0181
- T. Murashima, K. Nishi, K.-I. Nakamoto, A. Kato, R. Tamai, H. Uno, N. Ono: Preparation of Novel Heteroisoindoles from Nitropyridines and Nitropyridones, in: Heterocycles, 2002, 58, S. 301–310; doi:10.3987/COM-02-S(M)22.
- Joseph L. Duffy, Kenneth K. Laali: Aprotic Nitration (NO2+BF4−) of 2-Halo- and 2,6-Dihalopyridines and Transfer-Nitration Chemistry of Their N-Nitropyridinium Cations, in: J. Org. Chem., 1991, 56, S. 3006–3009; doi:10.1021/jo00009a015.
- O. Fischer: Notiz über Nikotinsäure aus Pyridin, in: Chem. Ber., 1882, 15, S. 62–64; doi:10.1002/cber.188201501180.
- E. F. Möller, L. Birkofer: Konstitutionsspezifität der Nicotinsäure als Wuchsstoff bei Proteus vulgaris und Streptobacterium plantarum , in: Chem. Ber., 1942, 75, S. 1108–1118; doi:10.1002/cber.19420750912.
- R. N. Shreve, E. H. Riechers, H. Rubenkoenig: Amination in the Heterocyclic Series by Sodium Amide, in: Ind. Eng. Chem., 1940, 32, S. 173–178; doi:10.1021/ie50362a008.
- G. M. Badger, W. H. F. Sasse: The action of metal catalysts on pyridines, in: Adv. Heterocyc. Chem., 1963, 2, S. 179–202; doi:10.1016/S0065-2725(08)60749-7.
- W. H. F. Sasse: 2,2'-Bipyridin In: Organic Syntheses. 46, 1966, S. 5–8, doi:10.15227/orgsyn.046.0005; Coll. Vol. 5, 1973, S. 102 (PDF).
- G. H. Burrows, L. A. King Jr.: The Free Energy Change that Accompanies Hydrogenation of Pyridine to Piperidine, in: J. Am. Chem. Soc., 1935, 57, S. 1789–1791; doi:10.1021/ja01313a011.
- J.D. Cox, G. Pilcher: Thermochemistry of Organic and Organometallic Compounds, Academic Press, New York, 1970, S. 1–636, ISBN 0-12-194350-X.
- D. D. Tanner, C.-M. Yang: On the structure and mechanism of formation of the Lansbury reagent, lithium tetrakis(N-dihydropyridyl)aluminate, in: J. Org. Chem., 1993, 58, S. 1840–1846; doi:10.1021/jo00059a041.
- A. J. De Koning, P. H. M. Budzelaar, J. Boersma, G. J. M. van der Kerk: Specific and selective reduction of aromatic nitrogen heterocycles with the bis-pyridine complexes of bis(1,4-dihydro-1-pyridyl)zinc and bis(1,4-dihydro-1-pyridyl)magnesium, in: J. Organomet. Chem., 1980, 199, S. 153–170; doi:10.1016/S0022-328X(00)83849-8.
- M. Ferles: Coll. Czech. Chem. Comm., 1959, 24, S. 1029–1033.
- Bericht zur Entwicklung der Pyridinproduktion in China (englisch).
- A Leading Provider in a Variety of Markets. In: vertellus.com. Vertellus Specialties Inc., abgerufen am 18. März 2016 (englisch).
- E. J. Corey, W. Suggs: Pyridinium Chlorochromate. An Efficient Reagent for Oxidation of Primary and Secondary Alcohols to Carbonyl Compounds, in: Tetrahedron Lett., 1975, 16, S. 2647–2650; doi:10.1016/S0040-4039(00)75204-X.
- C. H. Bamford, C. F. H Tipper: Comprehensive Chemical Kinetics: Non-radical Polymerisation, 1. Auflage, Elsevier, Amsterdam, 1980, ISBN 0-444-41252-2.
- A. V. Hopper: Recent Developments in Polymer Research, 1. Auflage, Nova Science Publisher, 2007, ISBN 1-60021-346-4.
- R. H. Crabtree: Iridium compounds in catalysis, in: Acc. Chem. Res, 1979, 12, S. 331–337; doi:10.1021/ar50141a005.
- Environmental Health Criteria (EHC) für Paraquat and Diquat, abgerufen am 19. November 2014.
- K. Eller, E. Henkes, R. Rossbacher, H. Hoke: Amines, Aliphatic, in: Ullmann's Encyclopedia of Industrial Chemistry, 2005, Wiley-VCH Weinheim.
- C. E. Terry, R. P. Ryan, S. S. Leffingwell: Toxicology Desk Reference: The Toxic Exposure & Medical Monitoring Index: The Toxic Exposure and Medical Monitoring Index, 5. Auflage, S. 1062, Taylor&Francis, ISBN 1-56032-795-2.
- A. Täufel, W. Ternes, L. Tunger, M. Zobel: Lebensmittel-Lexikon, 4. Auflage, S. 218, Behr Verlag, 2005, ISBN 3-89947-165-2.
- Uni Jena: Wasserbestimmung mit Karl-Fischer-Titration (Memento vom 4. März 2012 im Internet Archive).
- ECOTOX Database der Environmental Protection Agency (EPA).
- Datenblatt Pyridin bei AlfaAesar, abgerufen am 3. Juni 2010 (PDF) (JavaScript erforderlich).
- N. Bonnard, M.T. Brondeau, S. Miraval, F. Pillière, J.C. Protois, O. Schneider: Pyridine – Fiche toxicologique n° 85, INRS, 2011 (französisch).
- Yann Grosse, Dana Loomis, Kathryn Z Guyton, Fatiha El Ghissassi, Véronique Bouvard, Lamia Benbrahim-Tallaa, Heidi Mattock, Kurt Straif: Some chemicals that cause tumours of the urinary tract in rodents. In: The Lancet Oncology. 18, 2017, S. 1003–1004, doi:10.1016/S1470-2045(17)30505-3.
- G. Junk, C. Ford: A review of organic emissions from selected combustion processes, in: Chemosphere, 1980, 9, S. 187–230; doi:10.1016/0045-6535(80)90079-X.
- S. B. Hawthorne, R. E Sievers: Emissions of organic air pollutants from shale oil waste waters, in: Environ. Sci. Technol., 1984, 18, S. 483–490; doi:10.1021/es00124a016.
- D. H. Stuermer, D. J. Ng, C. J. Morris: Organic contaminants in groundwater near an underground coal gasification site in northeastern Wyoming, in: Environ. Sci. Technol., 1982, 16, S. 582–587; doi:10.1021/es00103a009.
- National Occupational Exposure Survey 1981–83, Cincinnati, OH, Department of Health and Human Services, Public Health Service, Centers for Disease Control, National Institute for Occupational Safety and Health.