Komplexchemie
Die Komplexchemie (lat. complexum, „umgeben“, „umarmt“, „umklammert“) ist ein Bereich der Anorganischen Chemie. Der Begriff Koordinationschemie wird im Allgemeinen synonym dazu verwendet. Die Komplexchemie befasst sich mit Komplexen bzw. Koordinationsverbindungen, die aus einem oder mehreren Zentralteilchen und einem oder mehreren Liganden aufgebaut sind. Die Zentralteilchen sind dabei meist Atome bzw. Ionen von Übergangsmetallen, die ungeladen oder geladen sein können.
Im Gegensatz zu herkömmlichen kovalenten Bindungen steuern in Komplexen gewöhnlicherweise die Liganden alle Elektronen zur Bindung bei, anstatt dass jeder Reaktionspartner mit jeweils einem Elektron zu einer Elektronenpaarbindung beiträgt (koordinative Bindung); trotzdem gibt es auch Komplexe eher kovalenter Natur.[1] Die Bildung von Komplexen lässt sich somit als Säure-Base-Reaktion im Sinne der Lewis-Definition verstehen, in der die Liganden (Basen) als Elektronenpaardonatoren auftreten und das Zentralteilchen (Säure) durch Lücken in seiner Elektronenkonfiguration als Akzeptor.
Komplexverbindungen spielen in verschiedenen Bereichen eine wichtige Rolle: In der Technik (zum Beispiel als Katalysator, siehe Abbildung des Grubbs-Katalysators rechts), in der Biologie (Hämoglobin und Chlorophyll), in der Medizin (Chelat-Therapie) oder in der Analytischen Chemie. Verbindungen mit organischen Liganden sind zudem Gegenstand der Metallorganischen Chemie. Da Komplexe von Übergangsmetallen mitunter sehr farbig sind, werden diese außerdem auch als Farbstoffe bzw. Pigmente eingesetzt (Berliner Blau). Besonders intensive Färbungen zeigen die Charge-Transfer-Komplexe, wie z. B. Permanganate.
Lange Zeit hatten Chemiker keine Vorstellung vom Aufbau koordinativer Verbindungen, die man als „Verbindungen höherer Ordnung“ bezeichnete. Zudem ließen sich viele Verhaltensweisen von Komplexen mit den damaligen Theorien nicht erklären, wie zum Beispiel die Stabilität von Cobalt(III)-chlorid in wässriger Lösung bei Zugabe von Ammoniak. Für die richtige Deutung der Struktur- und Bindungsverhältnisse in Komplexen erhielt der Schweizer Alfred Werner im Jahre 1913 als erster und jahrzehntelang einziger Anorganiker den Nobelpreis für Chemie.
Geschichte
.svg.png.webp)
Die systematische Erforschung der Struktur von Komplexverbindungen begann im späten 19. Jahrhundert und ist vor allem von den Namen Sophus Mads Jørgensen und Alfred Werner geprägt. Jørgensen machte sich zwar mit der Synthese zahlreicher neuer Komplexe einen großen Namen, war aber ein Anhänger der von Christian W. Blomstrand eingeführten „Kettentheorie“.[2] Nach dieser Theorie reihen sich die Liganden hintereinander und bilden somit Ketten, was heutzutage wenig sinnvoll wirkt, aber dafür immerhin die Wertigkeit des Metalls berücksichtigte.
Werner, der jahrzehntelang als Gegenspieler Jørgensens galt[3], formulierte 1892 hingegen eine Theorie, die grundsätzlich heute noch gültig ist, und im Folgejahr erstmals publiziert wurde.[4] Die daraus ableitbaren Aussagen etwa über mögliche Stereoisomerie konnten experimentell bestätigt werden und festigten die Theorie im Laufe der Jahre. Wenige Jahre nach der Auszeichnung mit dem Chemienobelpreis starb Werner 1919.
In den folgenden Jahrzehnten führten die Entwicklungen neuer Theorien über die chemische Bindung und neuer Technologien wie Röntgenstrukturanalyse zu großen Fortschritten in der Koordinationschemie. Zudem gab es seitdem weitere Chemienobelpreise auf dem Gebiet der Komplexchemie, beispielsweise 1973 für die Erforschung von sogenannten Sandwich-Komplexen durch Ernst Otto Fischer und Geoffrey Wilkinson, oder 2005 für die Beschreibung der Olefinmetathese und der Entwicklung geeigneter (komplexer) Katalysatoren durch Yves Chauvin, Robert Grubbs und Richard R. Schrock.
Nomenklatur
Komplexformeln
Die Oxidationszahl wird bestimmt, indem man die ursprüngliche Ladung des Zentralatoms betrachtet, als ob alle Liganden unter Mitnahme der gemeinsamen Elektronenpaare entfernt werden würden. Die Summe der Ladungsbeiträge der Liganden und der Oxidationszahl des/der Zentralteilchen muss die Ladung des Komplexes ergeben.
- Die Koordinationseinheit wird in eckigen Klammern dargestellt. Eine eventuell vorhandene Ladung wird als Exponent hinter der eckigen Klammer geschrieben. In diesem Fall wird die Koordinationseinheit auch als Komplex-Ion bezeichnet.[5]
- Das Zentralteilchen wird vor den Liganden genannt.
- Liganden werden (im Gegensatz zu früheren Empfehlungen, um Probleme mit sogenannten Non-innocent-Liganden zu vermeiden), unabhängig von ihrer Ladung, alphabetisch aufgeführt (Abkürzungen für Liganden werden ebenfalls in alphabetischer Reihenfolge angegeben, d. h. CH3CN vor MeCN vor NCMe).[6]
- Für mehratomige Liganden (sowie Abkürzungen für Liganden) werden runde Klammern verwendet. Weiterhin ist das Donoratom möglichst zuerst zu nennen (beispielsweise sind NCS- und SCN-Bindungsisomere).[6]
- Die Oxidationszahl kann als Exponent (römische Ziffer) am Zentralatom angegeben werden (das positive Vorzeichen wird vernachlässigt). Diese Angabe ist fakultativ.[6]
- In mehrkernigen Komplexen wird die Bindungsart von Brückenliganden mit dem griechischen Buchstaben μ bezeichnet. Dabei gibt ein Index n die Anzahl der verknüpften Zentralatome an. Bei Liganden mit mehreren Donoratomen wird die Haptizität mit dem griechischen Buchstaben η angegeben. Dahinter steht hochgestellt die Anzahl der Donoratome.
Komplexnamen
Bei der systematischen Benennung von Komplexsalzen gibt man zuerst das Kation und dann das Anion an, gleichgültig ob jeweils komplex oder nicht. Die Nennung der Bestandteile einer Koordinationseinheit geschieht in folgender Reihenfolge:
- Anzahl der Liganden: Ein mehrfaches Auftreten von Liganden wird durch griechische Zahlwörter (mono, di etc.) als Präfix angegeben. Bei Liganden mit komplexeren Namen oder zur Vermeidung von Mehrdeutigkeiten (z. B. dithiosulfat) verwendet man deren Multiplikativa (bis, tris etc.). Der hierdurch vervielfachte Teil kommt in Klammern.
- Art der Liganden: Die verschiedenen Liganden werden ohne Berücksichtigung ihrer Anzahl und ihrer Ladung in alphabetischer Reihenfolge genannt. Anionische Liganden erhalten die Endung „o“ an ihren Anionennamen (z. B. chlorido) und Radikale die Endung „-yl“ (z. B. nitrosyl). Die Namen neutraler oder kationischer Liganden werden nicht verändert. Ausnahmen von dieser Regel sind die Namen von Wasser (aqua), Ammoniak (ammin), CO (carbonyl) und NO (nitrosyl).[7]
- Koordinationseinheit: Bei einer anionischen Koordinationseinheit erhält das Zentralion die lateinische Schreibweise gefolgt von der Endung „at“ (z. B. Cuprat von lat. cuprum). Ist die Koordinationseinheit ein Kation oder neutral, wird der unveränderte deutsche Name verwendet.
- Ladung des Zentralions: Die Oxidationszahl des Zentralions(atoms) wird durch eine in runden Klammern gesetzte römische Ziffer angegeben und dem Namen der Koordinationseinheit nachgestellt. Ein Pluszeichen wird nicht geschrieben, für Null wird die arabische Ziffer benutzt.
Der vollständige Name der Koordinationseinheit wird in einem Wort geschrieben. Bis auf die Namen der Liganden aqua, ammin und nitrosyl werden die Namen aller neutraler Liganden in Klammern gesetzt. Die Namen anorganischer anionischer Liganden werden dann in runde Klammern gesetzt, wenn sie bereits numerische Vorsilben enthalten oder wenn dadurch Mehrdeutigkeiten vermieden werden. Im Namen von Komplexsalzen wird zwischen den Namen des Kations und des Anions ein Bindestrich geschrieben.
Aufbau von Komplexen
Zentralteilchen
Als Zentralteilchen kommen hauptsächlich Übergangsmetalle in Betracht, die über entsprechend freie d-Orbitale verfügen, mit denen sich die Liganden verbinden können. Ebenso häufig treten Komplexe auch bei Lanthanoiden und Actinoiden auf. Die Anzahl der Valenzelektronen beeinflusst maßgeblich Art, Stabilität und Aufbau der Komplexe, die gebildet werden. Erklärungen hierfür finden sich in entsprechenden Theorien, auf die noch eingegangen wird. Beispiele für Zentralteilchen sind:
Normalerweise haben die Metallteilchen in einem Komplex eine positive Oxidationszahl, allerdings können auch Verbindungen mit Metallatomen der Oxidationsstufe Null durch Reaktion von Metallen oder Metalldämpfen mit den entsprechenden Liganden hergestellt werden. Ein Beispiel ist die Reaktion von Nickel mit Kohlenstoffmonoxid zu Tetracarbonylnickel (Mond-Verfahren) oder von Eisen zu Pentacarbonyleisen. Derartige Reaktionen können auch zur Reinigung der entsprechenden Metalle genutzt werden (Chemischer Transport). Durch Reaktion von Metallen mit Liganden aus der Gasphase lassen sich beispielsweise Carbonyl-, Phosphin-, Olefin-, Aromat- und Cyclopentadienylkomplexe herstellen, wobei die Bildung gerade bei letzteren allerdings mit einer Redoxreaktion verbunden ist.[8]
Liganden
Komplexe können aus gleichen oder verschiedenen Liganden bestehen. Enthält ein Komplex ausschließlich gleichartige Liganden, so nennt man ihn homoleptisch, anderenfalls heteroleptisch. Da in Komplexen die Zahl der Partner in einer Bindung mit dem Zentralteilchen oft unabhängig von der Oxidationsstufe des Zentralteilchens ist, gibt es für Komplexe auch noch den aus dem 19. Jahrhundert stammenden Begriff „Verbindungen höherer Ordnung“.
Liganden können unterschiedliche Anzahlen an Elektronen in die Bindung einbringen, beispielsweise bringen Cl− und PPh3 zwei Elektronen mit ein, η5-Cp und η6-C6H6 sechs Elektronen, unverbrücktes μ1-CO (siehe Brücke (Chemie)) zwei Elektronen, verbrücktes μ2-CO ein Elektron (siehe Haptizität). Liganden können aber nicht nur unterschiedlich viele Elektronen einbringen, sondern bei entsprechender Größe auch mit mehreren Stellen gleichzeitig an demselben Zentralteilchen koordinieren. Die Anzahl der hierbei möglichen Bindungen wird Zähnigkeit genannt. Die gängigen einfachen Liganden wie Aqua (H2O), Ammin (NH3), Chlorido (Cl−) oder Cyanido (CN−) sind alle einzähnig bzw. monodentat, und binden z. B.: H3N—M.
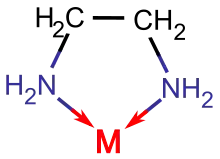
Liganden, die mehrere Koordinationsstellen für dasselbe Metallteilchen besitzen, bezeichnet man als Chelatliganden (griech. chelé, „Krebsschere“). Die dabei gebildeten Chelatkomplexe besitzen sowohl thermodynamisch als auch kinetisch eine höhere Stabilität. Die hohe thermodynamische Stabilität beruht auf der Erhöhung der Entropie des Systems, da zur Bildung eines beispielsweise oktaedrischen Komplexes mit einem zweizähnigen Liganden (Ligand mit zwei Koordinationsstellen) in wässriger Lösung folgende Reaktion abläuft:

Hier werden aus vier freien Teilchen (auf der linken Seite) sieben freie Teilchen (auf der rechten Seite). Die kinetische Stabilität beruht darauf, dass sich zur Bildung des Komplexes (nach der kinetischen Gastheorie) weniger Teilchen treffen müssen und bei der Dissoziation alle Bindungen eines Liganden zum Zentralteilchen gleichzeitig geöffnet werden müssen.
Gängige Chelatliganden sind unter anderem das zweizähnige Ethylendiamin (en), die vierzähnige Nitrilotriessigsäure (NTA) und das sechszähnige Ethylendiamintetraacetat (EDTA). Letzteres wird beispielsweise medizinisch dazu genutzt, giftige Schwermetalle wie Blei oder Quecksilber im Körper zu binden und anschließend auszuscheiden (Chelat-Therapie).
Anionische Liganden
Anionische Liganden werden aus dem Namen des Anions durch Anhängen der Endung -o gebildet: aus -id wird -ido, aus -it wird -ito und aus -at wird -ato. Einige traditionelle Namen (wie Chloro, Cyano oder Oxo) waren bis zur Revision der IUPAC-Nomenklatur im Jahr 2005 ebenfalls erlaubt.[9] Diese Ausnahmen sind durch die Überarbeitung nun weggefallen und nicht mehr zulässig.[10]
- H− (hydrido)
- F− (fluorido); Cl− (chlorido); Br− (bromido); I− (iodido)
- O2− (oxido);O22− (peroxido); OH− (hydroxido)
- Oxalat (ox)
- Acetylacetonat (acac)
- S2− (thio); SO42− (sulfato); S2O32− (thiosulfato)
- SCN− (thiocyanato bzw. isothiocyanato bei Koordination über N)
- CN− (cyanido, isocyanido bei Koordination über N bzw. cyano-C und cyano-N)
- NO2− (nitrito, nitro bei Koordination über N bzw. nitrito-N und nitrito-O); NO3− (nitrato)
- Ethylendiamintetraacetat EDTA4− (Ethylendiamintetraacetato)
- Nitrilotriessigsäure (NTA3−) (Nitrilotriacetato)
- Porphin/Porphyrine (bedeutsam in der Biochemie)
Neutrale Liganden
- NH3 (ammin)
- Ethylendiamin (en)
- H2O (aqua, veraltet aquo)
- CO (carbonyl)
- NO (nitrosyl)
Kationische Liganden
- NO+ (nitrosyl)
Organische Liganden
- Cyclopentadienyl-Anion (Cp)
Räumliche Gestalt von Komplexen (Molekülgeometrie)
Koordinationszahl und Koordinationspolyeder
4.svg.png.webp)
tetraedrischer Komplex
oktaedrischer Komplex
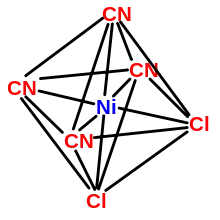
Die Koordinationszahl (KZ) gibt an, mit wie vielen Donoratomen der Liganden sich ein Zentralteilchen umgibt. Abhängig von der Koordinationszahl ordnen sich die Liganden in bestimmten Anordnungen um das Zentrum, die häufig, aber nicht immer, mit den Vorhersagen des VSEPR-Modells übereinstimmen. Denkt man sich zwischen den Liganden verbindende Linien, so erhält man die Koordinationspolyeder, mit denen die Struktur von Komplexen üblicherweise beschrieben wird. Gängig sind dabei Koordinationszahlen von 2 bis 9, darüber hinausgehende Zahlen können nur bei besonders großen Zentralteilchen und Chelatliganden erreicht werden. Am häufigsten sind allerdings die Koordinationszahlen 4 und 6. Nach dem VSEPR-Modell können folgende Polyeder angenommen werden:
- KZ 2: ein linearer Komplex, z. B. [AuCl2]− oder gewinkelter Komplex z. B. [Au(tBuXanthPhos)][AuBr2]
- KZ 3: eine trigonal-planare oder eine trigonal-aplanare Struktur (das Zentralteilchen liegt nicht exakt in der Mitte des Dreiecks, sondern leicht darüber)
- KZ 4: ein Tetraeder oder eine quadratisch-planare Struktur
- KZ 5: eine quadratisch-pyramidale oder trigonal-bipyramidale Struktur; beide sind durch die Berry-Pseudorotation ineinander überführbar und liegen bei entsprechender Temperatur im Gleichgewicht
- KZ 6: meist ein Oktaeder, teilweise ein trigonales Antiprisma oder (seltener) ein trigonales Prisma
- KZ 7 (sehr selten): eine pentagonale Bipyramide, ein einfach überkapptes Oktaeder oder ein einfach überkapptes trigonales Prisma
- KZ 8: ein quadratisches Antiprisma, ein trigonales Dodekaeder, ein zweifach überkapptes trigonales Prisma oder, seltener, ein Hexaeder (Würfel)
- KZ 9: ein dreifach überkapptes trigonales Prisma, z. B. [ReH9]2− oder überkapptes quadratisches Antiprisma z. B. [Ln(thf)[N(CH2CH2OH)3]]3+
- KZ 12: Ikosaeder oder ein Kuboktaeder ergibt z. B. [Ce(NO3)6]2- oder [Zr(η3–BH4)4]
Symmetrie
Da die Zentralteilchen und Liganden eines stabilen Komplexes genau wie die Ionen innerhalb von Kristallgittern geometrisch geordnete Strukturen einnehmen, werden sie bestimmten Punktgruppen zugewiesen. Die Kennzeichnung erfolgt üblicherweise nach der Schoenflies-Symbolik.
Isomerie
Wenn mehrere voneinander unterscheidbare Verbindungen für ein und dieselbe Summenformel existieren, spricht man (wie auch bei organischen Verbindungen) von Isomerie. Für Komplexverbindungen sind die Konfigurationsisomerie, Enantiomerie, Ionisations-Isomerie und die Bindungs-Isomerie relevant.
Bindungsisomerie
Die Bindungsisomerie kann auftreten, wenn ein Ligand mit unterschiedlichen Atomen an das Metallzentrum koordinieren kann. So kann etwa der Ligand SCN− sowohl mit dem Schwefelatom anbinden (thiocyanato) als auch mit dem Stickstoffatom (isothiocyanato).
Konfigurationisomerie
Je nach Koordinationszahl und Zusammensetzung einer Komplexverbindung, treten cis-trans-Isomerie bzw. faciale oder meridionale Anordnung auf.
cis-trans-Isomerie
Die cis-trans-Isomerie tritt bei quadratisch planaren (Koordinationszahl KZ = 4) und oktaedrischen (KZ = 6) Komplexen auf. In der nebenstehenden Abbildung dargestellt sind das cis- und das trans-Isomer einer oktaedrisch koordinierten Verbindung der allgemeinen Formel MA4BL, wobei L sowohl einen weiteren Liganden vom Typus B, als auch einen Liganden der dritten Sorte C darstellen kann. Für die Bezeichnung ist nur von Bedeutung, ob sich die beiden „besonderen“ Liganden (zweimal B bzw. einmal B und einmal C) zueinander (cis) oder voneinander weg (trans) anordnen. Entsprechend bilden die vier Liganden des Typs A eine Wippe (cis) bzw. eine quadratische Ebene (trans).
Wenn man in der Skizze die beiden A-Liganden, die nicht auf der Äquatorialebene liegen, gedanklich entfernt, erhält man einen quadratisch planaren Komplex mit der allgemeinen Formel MA2BL. Wie beim oktaedrischen Komplex ist von Belang, ob B und L zueinander (cis) oder voneinander weg (trans) weisen. Sobald im quadratisch planaren Komplex vier verschiedene Sorten von Liganden vorliegen, erhöht sich die Anzahl der möglichen Isomere auf drei.
fac- und mer-Anordnung
Die faciale (fac-) oder die meridionale (mer-)Anordnung tritt für oktaedrische Komplexe der allgemeinen Formel MA3B3 auf. Lassen sich die Liganden vom Typus A und die von Typus B durch eine Ebene, die das Zentralteilchen M enthält, klar trennen, weisen jeweils drei Liganden der fac-Anordnung in eine Richtung, wie zwei voneinander abgewandte Gesichter (engl. faces). Wenn sich die Liganden der beiden Sorten nicht voneinander abtrennen, sondern bloß auf orthogonale Ebenen der reinen Sorte A bzw. B aufteilen lassen, spricht man von der mer-Anordnung, da sich stets drei gleichartige Liganden auf dem Meridian der Kugel befinden müssen, deren Oberfläche alle sechs Liganden beinhaltet.
Enantiomerie
Die Voraussetzung für Enantiomerie ist Chiralität, wobei ein Metallkomplex auch ein chirales Metallzentrum aufweisen kann, ohne dass ein Ligand-Molekül selbst chiral ist. Je nach Art der Koordination bestehen verschiedene Anforderungen, die erfüllt sein müssen, damit optische Isomerie auftritt:
- tetraedrische (KZ = 4) Metallkomplexe sind chiral, wenn vier verschiedene Liganden an das Metallzentrum gebunden sind.
- quadratisch planare (KZ = 4) Metallkomplexe sind chiral, sobald sich sterisch anspruchsvolle Liganden gegenseitig behindern, und somit die Drehung um eine Ligand-Metall-Bindung verhindern.
- oktaedrische (KZ = 6) Metallkomplexe weisen häufig chirale Zentren auf, wenn sie Chelatliganden enthalten.
Bildung von Komplexen
Die klassische Komplexbildungsreaktion ist eine Säure-Base-Reaktion nach der Theorie von Gilbert Newton Lewis. Hierbei stellt das Zentralteilchen die Lewis-Säure (Elektronenpaar-Akzeptor) dar; der Ligand ist die Lewis-Base, also ein Molekül oder ein Ion, welches mindestens ein freies Elektronenpaar (Elektronenpaar-Donator) zum Ausbilden einer Bindung zur Verfügung stellen kann.
Ein Beispiel für eine typische Komplexbildung ist die Zugabe von Wasser zu Kupfer(II)-sulfat. Das farblose Salz reagiert mit dem Wasser zu einem blauen Komplex:

Dabei reagiert das Cu2+ als Lewis-Säure und das Wasser mit seinen freien Elektronenpaaren als Lewis-Base und es entsteht ein Hexaaquakomplex. Diese Reaktion wird aufgrund der gut sichtbaren Wirkung häufig im Chemieunterricht in der Schule als Nachweis für Wasser verwendet.
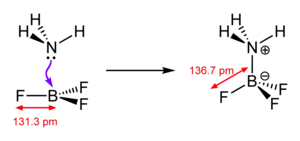
Die Art der chemischen Bindung, die aus der Bildungsreaktion resultiert, wird als koordinative Bindung (auch – veraltet[11] – als dative Bindung oder Donator-Akzeptor-Bindung) bezeichnet, und somit von den anderen Formen der chemischen Bindung (kovalente Bindung, Ionenbindung, Metallbindung) unterschieden. Diese (umstrittene) Unterscheidung wird damit gerechtfertigt, dass das bindende Elektronenpaar ursprünglich meist allein vom Liganden stammt, und nicht (wie in einer kovalenten Bindung) je ein Elektron von jedem Bindungspartner. In alten Lehrbüchern wird die Bindung teilweise noch durch einen Pfeil in Richtung des Akzeptors gekennzeichnet, allerdings sind diese Darstellungen veraltet. Eine koordinative Bindung wird heutzutage in Analogie zur kovalenten Bindung als Linie gezeichnet (siehe z. B. nebenstehende Skizze), denn ein Komplex ist zwar in der Regel als Lewis-Säure-Base-Adukt zu betrachten, die Koordination kann jedoch, wie etwa im Bereich der homogenen Katalyse, auch durch oxidative Addition erfolgen, wobei ein Teil der Bindungselektronen vom metallischen Zentralatom beigesteuert werden, welche im Zuge einer reduktiven Eliminierung wieder an diesem verbleiben können.
Ein typisches Beispiel für eine koordinative Bindung ist der in nebenstehender Skizze dargestellte Fall. Ammoniak (NH3) besitzt ein freies Elektronenpaar, das für eine koordinative Bindung zur Bildung des Moleküls H3N-BF3 zur Verfügung steht. Formal überträgt das Stickstoffatom hierbei ein Elektron an das Boratom, wodurch ersteres (allgemein: der Donor) eine formale positive, letzteres (allgemein: der Akzeptor) eine formal negative Ladung erhält. Man beachte, dass diese formalen Ladungen nichts mit der tatsächlichen Ladungsverteilung zu tun haben: Da Stickstoff eine wesentlich höhere Elektronegativität als Bor besitzt (3,0 gegenüber 2,0), ist die Bindung zum Stickstoff hin polarisiert (und die Oxidationsstufen bleiben unverändert).[12]
Darüber hinaus gibt es Komplexe, deren Bindungsverhältnisse sich erst durch anspruchsvollere Konzepte (etwa der Molekülorbitaltheorie) wirklich adäquat beschreiben lassen, wie etwa Metallcluster, Sandwichkomplexe (z. B. Bis(benzol)chrom, Manganocen und Ferrocen), die verwandten Halbsandwichverbindungen, aber auch Olefinkomplexe (wie das Zeise-Salz).
Bindungsverhältnisse
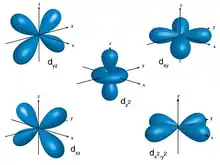
Die Bindung zwischen Zentralteilchen und Liganden und die Stabilität von Komplexen kann durch unterschiedliche Modelle genauer beschrieben werden, die es ermöglichen, auch Aussagen über Eigenschaften wie Farbigkeit oder Magnetismus zu treffen.
VB-Theorie
Die früheste Erklärung lieferte die Valenzstrukturtheorie (valence bond theory, VB-Theorie). Diese nimmt an, dass besetzte Ligandenorbitale mit unbesetzten Orbitalen des Zentralteilchens überlappen und somit eine Bindung ausbilden. Um die räumliche Struktur von Komplexen zu erklären, wird von der Entstehung von Hybridorbitalen beim Metall ausgegangen. Dadurch erklärt die VB-Theorie zwar die Geometrie und magnetische Eigenschaften, nicht jedoch beispielsweise die Farbigkeit. Mit der 18-Elektronen-Regel kann zudem in bestimmten Fällen die Stabilität von Übergangsmetallkomplexen abgeschätzt werden, wobei der Geltungsbereich der Regel allerdings stark beschränkt ist.
Kristall- und Ligandenfeldtheorie
Als Weiterentwicklung gilt die Kristallfeldtheorie, die von reinen elektrostatischen Wechselwirkungen zwischen den Liganden und dem Zentralteilchen ausgeht und auch die Farbigkeit der Komplexe erklären kann. Das Modell wird im Sprachgebrauch häufig mit der Ligandenfeldtheorie vermischt, die die Kristallfeldtheorie erweitert und den Einfluss der punktförmigen Liganden auf die Energien der d-Orbitale des Zentralmetalls untersucht. Diese Betrachtungsweise ist aufgrund ihres Vermögens, trotz ihrer Einfachheit viele Eigenschaften erklären zu können, heute noch sehr gängig.
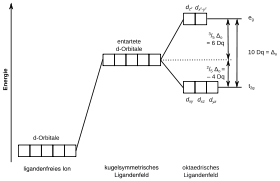
Die Energien der d-Orbitale eines Metalls sind zunächst einmal entartet (energiegleich). Nähert sich nun ein kugelsymmetrisches Ligandenfeld an, erhöht sich die Energie aller d-Orbitale in gleichem Maße. Die Ligandenfeldtheorie betrachtet nun die räumliche Gestalt dieser Orbitale und die Geometrie des Ligandenfelds in Form von Punktladungen. Schaut man sich beispielsweise einen oktaedrischen Komplex an, so stellt man fest, dass in einem oktaedrischen Ligandenfeld aufgrund der Geometrie die dz2- und dx2−y2-Orbitale energetisch ungünstiger gelegen sind, und die Orbitale dxy, dyz und dxz wiederum günstiger. Die Summe dieser Energiedifferenzen wird Ligandenfeldstabilisierungsenergie (LFSE) genannt und in einem oktaedrischen Komplex mit ΔO bezeichnet. Der absolute Wert dieser Aufspaltung lässt sich experimentell durch Spektroskopie bestimmen und ist sowohl vom Zentrum als auch von den Liganden abhängig, wird aber grundsätzlich relativ als 10 Dq angegeben. Der Einfluss der Zentren und Liganden auf die Aufspaltung kann an der spektrochemischen Reihe abgelesen werden.
Die Energieerhöhung macht dabei 3/5 der LFSE aus und die Absenkung entsprechend 2/5. Werden alle Orbitale besetzt, gelangt man in der Summe zu 0 Dq, da sich der Energieschwerpunkt der d-Orbitale nicht ändern darf (gestrichelte Linie in der Abbildung). Die energetisch erhöhten Orbitale werden als eg-Orbitale bezeichnet (e für zweifach „entartet“) und die abgesenkten Orbitale als t2 g-Orbitale (t für „tripel-entartet“).[13] In einem Komplex werden diese Orbitale nun entsprechend der Hund-Regel mit Elektronen besetzt. In Abweichung dazu beobachtet man bei hohen Feldaufspaltungen auch die Bildung von Elektronenpaaren anstatt der Besetzung mit ungepaarten Elektronen, sofern die LFSE höher ist als die Spinpaarungsenergie. Solche Komplexe werden low-spin-Komplexe genannt, in Abgrenzung zu den üblichen high-spin-Komplexen.
Ähnliche Überlegungen lassen sich auch für andere Koordinationspolyeder als dem Oktaeder aufstellen. Damit kann die Ligandenfeldtheorie einfache Erklärungen unter anderem für das magnetische Verhalten von Komplexen liefern, indem die Paarung von Elektronen betrachtet wird, oder auch für die Farbe, die mit Elektronenübergängen zwischen den Orbitalen erklärt werden kann. Auch die geometrische Verzerrung durch den Jahn-Teller-Effekt kann hier anhand der Elektronenkonfiguration erklärt werden.
Spektrochemische Reihe
Die experimentell aufgestellte spektrochemische Reihe ordnet Liganden und Metallteilchen nach der Stärke der von ihnen verursachten Ligandenfeldaufspaltung. Bei den Liganden ergibt sich demnach die folgende Reihenfolge:
- I− < Br− < Cl− < F− < OH− < H2O < NC− < NH3 < CN− < CO
In dieser Reihe verursacht der Iodido-Ligand die kleinste Aufspaltung und der Carbonyl-Ligand die größte. Diese Aufstellung entspricht im Prinzip der Basenstärke nach dem Lewis-Konzept. Auf ähnliche Weise kann man auch die Metallteilchen sortieren:
- Mn2+ < Ni2+ < Co2+ < Fe2+ < V2+ < Fe3+ < Cr3+ < V3+ < Co3+ < Mn4+ < Mo3+ < Rh3+ < Pd4+ < Ir3+ < Re4+ < Pt4+
Hieraus wird die Faustregel deutlich, dass höhere Ionenladungen auch eine höhere Aufspaltung bewirken. Je weiter rechts ein Teilchen steht, umso wahrscheinlicher wird daher auch eine low-spin-Konfiguration in einem entsprechenden Komplex.
MO-Theorie
Die besten Ergebnisse liefert jedoch die Molekülorbitaltheorie, von der die Ligandenfeldtheorie nur ein Ausschnitt ist. Sie behandelt sowohl das Zentralteilchen als auch die Liganden quantenmechanisch und ist damit am genauesten, aber auch am anspruchsvollsten.
Stabilität
Zur Abschätzung oder Erklärung der Stabilität von Komplexverbindungen können mehrere Überlegungen in Betracht gezogen werden.
HSAB-Konzept
Das Prinzip der harten und weichen Säuren und Basen nach Ralph G. Pearson besagt, dass harte Säuren bevorzugt mit harten Basen reagieren und weiche Säuren entsprechend mit weichen Basen. Als hart werden Teilchen mit einer hohen Ladungsdichte bezeichnet und als weich solche mit geringer Ladungsdichte, die leicht polarisierbar sind. Dieses Konzept lässt sich auch auf die Stabilität von Komplexverbindungen anwenden.
Hin- und Rückbindung

Die MO-Theorie gibt weitere Aufschlüsse über die Stabilität beispielsweise von Metallcarbonylen. Demnach dienen in Komplexen alle Liganden als σ-Donoren, starke Liganden wie CO sind jedoch zusätzlich starke π-Akzeptoren. Die σ-Hinbindung geschieht über das HOMO des CO, das mit einem leeren d-Orbital des Metalls überlappt. Zusätzlich kann aber das LUMO des CO mit einem besetzten d-Orbital des Metalls geeigneter Symmetrie überlappen und bewerkstelligt damit eine π-Rückbindung, die dem Komplex zu besonderer Stärke verhilft. Zudem verstärkt diese Rückbindung wiederum die σ-Hinbindung, weshalb man hier von einer Synergie spricht.
Anwendung des Massenwirkungsgesetzes
Zur quantitativen Beschreibung der Stabilität von Komplexen lassen sich Gleichgewichtskonstanten aufstellen, da die Lewis-Säure-Base-Reaktionen zur Komplexbildung Gleichgewichtsreaktionen sind, für die das Massenwirkungsgesetz aufgestellt werden kann. Die Gesamtreaktion kann in einzelne Schritte unterteilt werden (sog. Elementarreaktionen), d. h. jeweils für die Anlagerung eines Liganden. Das Produkt der Gleichgewichtskonstanten der einzelnen Elementarreaktionen zur Komplexbildung ergibt dann die Gleichgewichtskonstante für die Gesamtreaktion.

Die resultierende Konstante nennt man Komplexbildungskonstante. Diese Konstante gibt auch an, wie stabil der Komplex ist bzw. ob er zur Dissoziation neigt. Daher wird die Komplexbildungskonstante auch Komplexstabilitätskonstante oder Komplexassoziationskonstante KA genannt. Ihr reziproker Wert wird als Komplexdissoziationskonstante KD bezeichnet, also KD = KA−1. Je höher die Komplexbildungskonstante KA, desto stabiler der Komplex, je kleiner, desto leichter ist die Dissoziation.
Komplexbildungskonstanten
Nach Martell & Smith, 1982; Hyvönen & Aksela, 2010; Vasilev, et al. 1996; Vasilev, et al. 1998[14]
| Komplexbildner | Abkürzung | Komplexbildungskonstante gegen Ca2+ |
|---|---|---|
| Diethylentriaminpentaessigsäure | DTPA | 10,8 |
| Ethylendiamintetraessigsäure | EDTA | 10,7 |
| β-Alanindiessigsäure | ADA | 5–7 |
| Methylglycindiessigsäure | MGDA | 7 |
| Nitrilotriessigsäure | NTA | 6,4 |
| Nitrilotrimethylenphosphonat | NTMP | 5,75 |
| Tetranatriumiminodisuccinat | IDS | 5,2 |
| Tetranatrium-N,N-bis(carboxylatomethyl)-L-glutamat | GLDA | 5,2 |
| Ethylendiamindibernsteinsäure | EDDS | 4,6 |
Spezielle Komplexverbindungen
Sandwich-Komplexe
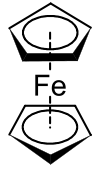
In Sandwichkomplexen sind die Metallzentren von zwei planaren und zyklischen organischen Liganden eingeschlossen wie von zwei Brötchenhälften, weshalb man dieser Art von Verbindung den besagten Namen gab. Zu den Sandwichkomplexen zählen unter anderem die Metallocene und die „Klavierstuhl-Komplexe“. Der gängigste Ligand ist dabei das Cyclopentadienyl-Anion (Cp), das sechs π-Elektronen besitzt und somit aromatisch ist (siehe Hückel-Regel). Es sind aber auch entsprechende Komplexe mit Benzol als Ligand möglich, beispielsweise Bis(benzol)chrom, oder das Uranocen mit Cyclooctatetraenyl-Liganden.
Der erste synthetisierte Sandwichkomplex war 1951 das Ferrocen, dessen mögliche Struktur aber einige Zeit lang Rätsel aufwarf. Ernst Otto Fischer, Geoffrey Wilkinson und Robert B. Woodward klärten die korrekte Struktur schließlich unabhängig voneinander auf, wofür die beiden ersteren 1973 den Chemienobelpreis erhielten. Ferrocen war bei der Forschung an Katalysatoren zufällig entdeckt worden und fiel durch die ungewöhnliche Stabilität seiner orangefarbenen Kristalle auf. Durch die Erfüllung der 18-Elektronen-Regel ist es stabiler als ähnliche Verbindungen mit anderen Metallen, wie Cobaltocen oder Nickelocen.
Die organischen Liganden binden bei dieser Art von Verbindungen mit ihren π-Elektronen an das Metallzentrum an. Da dies nicht unbedingt mit dem gesamten Ring passieren muss, sondern auch andere Bindungszustände möglich sind, gibt es zur Beschreibung dieser Verhältnisse den Begriff der Haptizität. Die Haptizität η beim Ferrocen beträgt beispielsweise 5, da jeder Ligand mit fünf Atomen anbindet. Dies schlägt sich auch in der Schreibweise [Fe(η5-C5H5)2] für den Komplex nieder.
Mehrkernige Komplexe
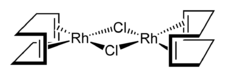
Mehrkernige Komplexe enthalten mehr als ein Zentralteilchen. Sie sind über einen Brückenliganden, wie beispielsweise Sauerstoff (O2−, OH−, H2O, OR) oder Chlor verbunden. Es gibt jedoch auch Komplexverbindungen mit (z. T. nicht-ganzzahligen) Metall-Metall Mehrfachbindungen, z. B. [Tc2X9]3−, X=Cl, Br.
Makrocyclische Metallkomplexe
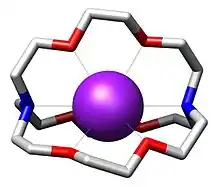
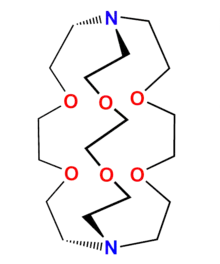
Bestimmte natürliche Antibiotika, die zum Typ der Cyclopeptide gehören (z. B. Valinomycin), sind in der Lage, selektiv Kaliumionen zu binden und zu transportieren. 1967 synthetisierte Charles Pedersen erstmals Kronenether, die zum Typ der makrocyclischen Polyether gehören und die in der Lage sind, insbesondere Alkali- und Erdalkaliionen zu komplexieren und zu transportieren. Ausgehend von diesen makrocyclischen Polyethern synthetisierte die Arbeitsgruppe von Jean-Marie Lehn 1969 erstmals einen makrobicyclischen Liganden (Azopolyether), der als Kryptand bezeichnet wurde. Dieser komplexierte ebenfalls in seinem Hohlraum Alkali- und Erdalkaliionen, die entsprechenden Komplexe wurden als Kryptate bezeichnet. In der Folge wurden verschiedene Kryptande hergestellt, die unterschiedlich große Hohlräume aufweisen und damit für die Größe von Alkali- bzw. Erdalkaliionen angepasst sind. Die Stabilitätskonstanten der entsprechenden Kryptate sind relativ hoch, die Komplexe weisen eine gute Selektivität für die Ionen auf und eignen sich daher zur selektiven Abtrennung der Ionen aus Lösungen. Ebenfalls gelang es in der Folge, makrotricyclische Kryptande und solche mit anderen Heteroatomen herzustellen.[15] Viele makrocyclische Metallkomplexe haben auch eine biologische Bedeutung. Weitere Beispiele sind Komplexe mit Phthalocyanin als Ligand, wie im Farbstoff Kupferphthalocyanin.
Gitterförmige Metallkomplexe
Gitterförmige Metallkomplexe sind supramolekulare Komplexverbindungen aus mehreren Metallatomen und koordinierenden Chelatliganden, die ein gitterförmiges Strukturmotiv ausbilden. Die Strukturbildung entsteht dabei meist über thermodynamische Selbstorganisation. Sie weisen Eigenschaften auf, die sie für die Informationstechnologie als zukünftige Speichermaterialien interessant machen.[16]
Anwendung
Biologische Bedeutung
In der Biologie spielen Komplexe eine wichtige Rolle. Es kann sich dabei um katalytisch aktive Proteine (Enzyme) oder katalytisch nicht aktive Proteine handeln. Zahlreiche Enzyme enthalten Komplexe in ihren aktiven Zentren. Dieses Thema ist eines der Schwerpunktgebiete der bioanorganischen Chemie. Im Allgemeinen liegt hierbei ein komplexierendes Metallatom vor, welches nicht vollständig durch Aminosäureseitenketten als Liganden komplexiert ist. Eine Ligandenstelle fungiert als aktives Zentrum zur Umsetzung oder temporären Bindung des Substrats. Häufigste Komplexzentren sind dabei Eisen, Kupfer, Zink, Calcium, Magnesium und Mangan. Es kommen aber auch ungewöhnlichere Elemente wie Vanadium vor. Insbesondere Calcium- wie auch Zink-Komplexe haben eine strukturelle Bedeutung (z. B. Zinkfinger bei der DNA-Sequenzerkennung).
Bei den katalytisch nicht aktiven Proteinen finden sich z. B. Porphyrinkomplexe wie das Häm im Hämoglobin und in Cytochromen, oder das Chlorophyll (jeweils Chelatkomplexe). Koordinationsverbindungen sind somit dafür verantwortlich, dass Blut rot erscheint und ein Blatt einer Pflanze grün.
Technik
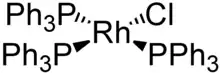
Komplexe finden in vielen chemischen Reaktionen Anwendung als Katalysatoren. Beispielsweise werden bei der bereits erwähnten Olefinmetathese, für die es 2005 einen Nobelpreis gab, Carbenkomplexe mit Ruthenium oder Molybdän verwendet (siehe Grubbs-Katalysator). Der Wilkinson-Katalysator ist ein quadratisch-planarer Rhodium(I)-Komplex, der für diverse Anwendungen wie die Hydrierung von Olefinen geeignet ist. Erwähnenswert ist auch die industrielle Herstellung von Essigsäure aus Methanol und Kohlenstoffmonoxid mit einem Rhodium-Katalysator im Monsanto-Prozess.
Diverse Komplexbildner dienen als Lebensmittelzusatzstoffe, als Additiv in der Wasch- und Reinigungsmittelindustrie, in der Galvano- und Leiterplattenindustrie sowie in der chemischen Analytik.
Phthalocyanin-Komplexe werden in CDs als Speichermedium verwendet.
In der analogen Fotografie wird nach der Entwicklung das verbliebene, unbelichtete, in Wasser kaum lösliche Silberbromid mit Fixiersalz-Lösung (Ammonium- oder Natriumthiosulfat) aus der Schicht gelöst: siehe Fixieren (Fotografie).
Forschung
Es ist grundsätzlich ein Problem, kurzlebige und instabile Moleküle, die bei Reaktionen als Zwischenprodukte auftreten, zu fixieren. Eine Methode ist die Fixierung durch Komplexbildung. Die fixierten Moleküle haben dabei allerdings andere chemische Eigenschaften, auf diese Art lassen sich aber Bindungs- und Strukturverhältnisse untersuchen. Beispiele hierfür sind Komplexe mit Carbenen, Cyclobutadien, Diiminen und carbenanalogen Silylenen. Nach Freisetzung aus den Komplexen sind die Moleküle wieder hoch reaktiv. Verwendet werden Metallkomplexe mit Chrom, Nickel, Eisen und Mangan. Als Ausgangskomplex wird oftmals ein Metallcarbonylkomplex verwendet. Beispiele: Tricarbonyl-cyclobutadieneisen, Methoxyphenyl-carben-pentacarbonylchrom, Tetrachlor-bis(tetramethylcyclobutadien)nickel.[17]
Siehe auch
Literatur
- Lutz H. Gade: Koordinationschemie. 1. Aufl., Wiley-VCH, Weinheim 1998, ISBN 978-3-527-29503-6.
- Christoph Janiak: Komplex-/Koordinationschemie, in: Erwin Riedel (Hrsg.): Moderne Anorganische Chemie. 3. Aufl., de Gruyter, Berlin 2007, S. 381–579, ISBN 978-3-11-019060-1.
- A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 102. Auflage. Walter de Gruyter, Berlin 2007, ISBN 978-3-11-017770-1, S. 1315–1400.
- Henry Taube: Elektronenübertragung zwischen Metallkomplexen – ein Rückblick (Nobel-Vortrag), in: Angewandte Chemie 1984, 96, S. 315–326, doi:10.1002/ange.19840960504.
Weblinks
Einzelnachweise
- A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 102. Auflage. Walter de Gruyter, Berlin 2007, ISBN 978-3-11-017770-1, S. 1316.
- William H. Brock: Viewegs Geschichte der Chemie, Vieweg, Braunschweig 1997, S. 364.
- Lutz H. Gade: Koordinationschemie. 1. Aufl., Wiley-VCH, Weinheim 1998, S. 6.
- Alfred Werner: Beitrag zur Konstitution anorganischer Verbindungen. in: Zeitschrift für Anorganische Chemie 1893, 3, S. 267–330, doi:10.1002/zaac.18930030136.
- Christoph Janiak: Komplex-/Koordinationschemie, in: Erwin Riedel (Hrsg.): Moderne Anorganische Chemie. 3. Aufl., de Gruyter, Berlin 2007.
- Nomenclature of Inorganic Chemistry, IUPAC Recommendations 2005, RSC Publishing, Cambridge, UK.
- IUPAC Red Book 2005, s. Section IR-9.2.2.3 für Komplexe (PDF-Datei; 4,1 MB).
- Max Herberhold: Komplexchemie mit nackten Metallatomen. In: Chemie in unserer Zeit. Band 10, Nr. 4, 1976, S. 120–129, doi:10.1002/ciuz.19760100405.
- Wolfgang Liebscher, Ekkehard Fluck: Die systematische Nomenklatur der anorganischen Chemie. Springer-Verlag, 1998, ISBN 978-3-540-63097-5, S. 127–150 (eingeschränkte Vorschau in der Google-Buchsuche).
- Karl-Heinz Hellwich: Mehr Systematik: Nomenclature of Inorganic Chemistry. Hrsg. von der International Union of Pure and Applied Chemistry. RSC Publishing, Cambridge/UK, 2005. XII + 366 S., geb., 49,95 . ISBN 0-85404-438-8. In: Nachrichten aus der Chemie. 54, 2006, S. 807–808, doi:10.1002/nadc.20060540725.
- Eintrag zu coordination. In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.C01329.
- Erwin Riedel: Allgemeine und Anorganische Chemie, Walter de Gruyter Verlag (1999), Abschnitt 2.2.3, ISBN 3-11-016415-9.
- Charles E. Mortimer und Ulrich Müller: Chemie. 10. Aufl., Thieme, Stuttgart 2010, S. 523.
- Dorota Kołodyńska: Chelating Agents of a New Generation as an Alternative to Conventional Chelators for Heavy Metal Ions Removal from Different Waste Waters S. 341, 345 doi:10.5772/21180
- Bernard Dietrich, Jean-Marie Lehn, Jean-Marie Sauvage: Kryptate: makrocyclische Metallkomplexe. In: Chemie in unserer Zeit. Band 7, Nr. 4, 1973, S. 120–128, doi:10.1002/ciuz.19730070405.
- J.-M. Lehn et al., Angew. Chem., 2004, 116, S. 3728–3747.
- Günter Schmid: Die Fixierung kurzlebiger Moleküle durch Komplexbildung. In: Chemie in unserer Zeit. Band 8, Nr. 1, 1974, S. 26–30, doi:10.1002/ciuz.19740080105.