Ataxie
Ataxie (aus griechisch ἀταξία ataxia ‚Unordnung‘ ‚Unregelmäßigkeit‘) ist in der Medizin ein Oberbegriff für verschiedene Störungen der Bewegungskoordination. Eine Ataxie kann auftreten, auch wenn keine Lähmung (Parese) vorliegt, also bei normaler Muskelkraft.
| Klassifikation nach ICD-10 | |
|---|---|
| R27.0 | Ataxie, nicht näher bezeichnet |
| ICD-10 online (WHO-Version 2019) | |
Formen der Ataxie
Als Rumpfataxie bezeichnet man die Unfähigkeit, gerade zu sitzen, so dass die Betroffenen nur noch mit Hilfe einer Stütze sitzen oder stehen können. Analog bezeichnet man als Standataxie die Unfähigkeit zu stehen, so dass die Betroffenen nur noch mit Hilfe steh- und gehfähig sind.
Menschen mit einer Gangataxie haben ein breitbeinig-unsicheres Gangbild. Eine Ataxie bei Zielbewegungen (auch afferente Ataxie genannt) führt zu Bewegungen falschen Ausmaßes mit Daneben-Zeigen (Dysmetrie), zu überschießend-ausfahrenden Bewegungen (Hypermetrie) oder zu unflüssig-verwackelten Bewegungen (Asynergie) und damit zur Unfähigkeit einer raschen Folge antagonistischer Bewegungen (Dysdiadochokinese).
Ursachen von Ataxien
Jede Schädigung der an der Bewegungssteuerung beteiligten Teilsysteme des Nervensystems kann eine Ataxie hervorrufen. So können neben verschiedenen degenerativen Erkrankungen, Entzündungen, Tumoren und Verletzungen auch Durchblutungsstörungen oder Stoffwechselstörungen zu Ataxie führen, sowie Vergiftungen, z. B. durch Kohlenmonoxid.
Häufigste Ursache von Ataxien sind Erkrankungen des Kleinhirns. Dieses ist für die Koordination der sensiblen Informationen aus dem Rückenmark, der Informationen des Gleichgewichtsorgans und der übrigen Sinneseindrücke und deren Umsetzung in motorische Bewegungsabläufe zuständig, also die Planung, Koordination und Feinabstimmung von Bewegungen.
Ataxien können deswegen auch auftreten, wenn die über das Rückenmark ankommenden sensiblen Informationen aus den peripheren sensiblen Nerven, Gelenken und Muskeln fehlen. Dadurch fehlt die für gezielte motorische Bewegungen erforderliche Feinsteuerung, es kommt zur sogenannten sensiblen Ataxie.
Gangataxie tritt auch auf beim Hydrocephalus normalis, auch altersbedingter Hydrozephalus oder Normaldruckhydrozephalus genannt. Hierbei wird das produzierte Hirnwasser (Liquor) nicht mehr in ausreichender Menge resorbiert oder in den Ventrikeln verteilt, es kommt zu einem zeitweisen Überdruck im Gehirn, dieser beeinflusst u. a. die Nerven, die die Bewegung steuern. Neben der Gangataxie treten häufig noch parallel Harndrang und eine Verringerung der kognitiven Leistungsfähigkeit, auch reversible Demenz genannt, auf. Das gleichzeitige Auftreten dieser drei Symptome nennt man Hakim-Trias.
Auch an sekundäre Ataxien infolge der Kupfer-Stoffwechselerkrankung Morbus Wilson sollte differentialdiagnostisch gedacht werden.
Zerebelläre Ataxie
Kleinhirn-Durchblutungsstörungen oder Kleinhirn-Blutungen, also Schlaganfälle des Kleinhirns, lösen eine Ataxie aus.
Ataxien treten nicht selten im Verlauf entzündlicher Erkrankungen des Nervensystems mit Schädigung des Kleinhirns und/oder Rückenmarks auf. Zu den wichtigsten entzündlichen Ursachen der zereberellären Ataxie gehören die multiplen Sklerose, paraneoplastische und nicht-paraneoplastische Antikörper-assoziierte Autoimmunerkrankungen[1][2][3] sowie infektiöse oder postinfektiöse Ataxien (z. B., neben vielen anderen, Zoster-Zerebellitis, Kleinhirn-Abszess, selten Borreliose nach Zeckenbiss).
Hirntumoren oder Metastasen im Kleinhirn und eine Einklemmung des Kleinhirns im Rahmen eines diffus erhöhten Hirndrucks jeder Ursache führen zur Ataxie, sofern nicht zuvor eine Bewusstseinstrübung eintritt.
Ataxien können durch chronische Vergiftungen ausgelöst werden, beim Menschen am häufigsten durch eine akute Kleinhirnfunktionsstörung im Rahmen eines akuten Alkoholrausches (reversibel) oder durch einen chronischen Alkoholismus (schlechtere Besserungstendenz). Als Symptom einer Überdosierung von Medikamenten kann es zu in der Regel schnell reversiblen Ataxien kommen, vor allem bei Überdosierung oder zu schneller Aufdosierung von Antiepileptika, ebenso als häufige Nebenwirkung von Medikamenten aus der Gruppe der Benzodiazepine, auch in geringer Menge, welche aber nach dem Absetzen reversibel ist. Ataxien durch Vergiftungen mit Schwermetallen (besonders Quecksilber) oder Pestiziden sind extrem selten.
Ferner können genetische Erkrankungen mit einer zerebellären Ataxie einhergehen (z. B. spinozerebelläre Ataxien), beispielsweise beim Richards-Rundle-Syndrom oder dem Gillespie-Syndrom.
Gluten-Ataxie
Gluten-Ataxie ist eine Autoimmunerkrankung, die durch den Verzehr von Gluten ausgelöst wird,[4][5] und ein interindividuell variables, progressives Beschwerdebild aufweist,[6] das von leichter Asynergie bis zu ausgeprägten unwillkürlichen Bewegungen reicht.[7] Frühe Diagnostizierung und Behandlung durch glutenfreie Ernährung kann die Beschwerden lindern und ein Voranschreiten verhindern. Die Effektivität der Behandlung hängt von der Zeit seit dem Einsetzen der Ataxie ab, weil das pathogenetische Absterben der Purkinjezellen in der Kleinhirnrinde irreversibel ist.[4][8] Gluten-Ataxien machen etwa 40 % der Ataxien von unbekannter Ursache und etwa 15 % aller Ataxien aus.[8] Weniger als 10 % der Betroffenen haben gastroenterologische Symptome, aber bei 40 % lässt sich eine Schädigung des Darms nachweisen.[4][8] Antikörper gegen Transglutaminase 6 können als Biomarker in der Diagnostik nützlich sein.[9] Ihr Spiegel fällt bei glutenfreier Diät erheblich, teils unter die Detektionsschwelle ab.[9]
Spinale oder sensible Ataxie
Auch hier ist die häufigste Ursache eine chronische Alkoholvergiftung. Ähnlich wie das Kleinhirn wird auch das Rückenmark häufig von der multiplen Sklerose betroffen. Tumoren im Rückenmark oder in der Wirbelsäule sind insgesamt selten. Eine sensible Ataxie wird häufiger durch eine in den Rückenmarksraum eingebrochene Wirbelkörper-Metastase hervorgerufen. In der Tiermedizin wird sie als Wobbler-Syndrom bezeichnet.
Eine nicht ganz seltene Ursache einer sensiblen Ataxie besteht in einer Schädigung der sensiblen Rückenmarksbahnen durch einen Vitamin-B12-Mangel (die sogenannte Funikuläre Myelose). Diese Schädigung ist bei entsprechender Behandlung komplett reversibel.
Weitere Vitamin-B-Mangelerkrankungen, die zu Ataxien führen können, sind Beriberi (B1-/Thiaminmangel) und in geringerem Maße Pellagra (B3-/Niacinmangel), durch Mangel- oder Fehlernährung (z. B. in Entwicklungsländern oder durch Vernachlässigung der Nahrungsaufnahme bei Anorexia nervosa u. ä. und Alkoholismus).
Als wichtige infektiöse Ursachen der sensiblen Ataxie sind die Syphilis des Nervensystems zu erwähnen, ferner auch HIV und die Infektion mit dem Zoster-Virus.
Hereditäre Ataxien
Ataxien treten ferner bei zahlreichen, insgesamt aber seltenen, genetisch bedingten (= hereditären) Erkrankungen auf, den hereditären Ataxien oder Heredoataxien.[10] Diese werden autosomal-rezessiv oder autosomal-dominant vererbt (ADCA = autosomal-dominante zerebellare Ataxien). Die autosomal-dominanten Ataxien werden heute zumeist spinocerebellare Ataxien (SCAs) genannt. Insgesamt 28 chromosomale Lokalisationen für SCAs sind bekannt, von denen ungefähr die Hälfte kloniert ist. Gemeinsam ist diesen Erkrankungen, dass sie das Kleinhirn und/oder die Hinterstränge des Rückenmarks befallen.
Autosomal-rezessive Ataxien
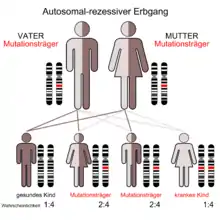
- Friedreich-Ataxie
- Ataxia teleangiectatica
- Vitamin-E-Mangel-Ataxie
- Morbus Refsum
- Abetalipoproteinämie (Bassen-Kornzweig-Syndrom)
- Metachromatische Leukodystrophie
- Adrenoleukodystrophie
- GM-Gangliosidose
- Früh beginnende zerebellare Ataxie mit erhaltenen Muskeleigenreflexen
- Saldino-Mainzer-Syndrom
- ARSAL
Autosomal-dominante zerebellare Ataxien (ADCA)
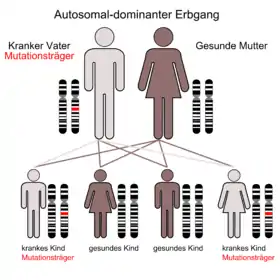
Die Gruppe der Autosomal-dominanten zerebellaren Ataxien umfasst eine Vielzahl an Erkrankungen:
- Spinozerebellare Ataxien (SCA) Typ 1–7
- Spinozerebellare Ataxie Typ 13
- Dentato-rubro-pallido-luysianische Atrophie mit Demenz
- ADCA mit pigmentärer Retinadegeneration (ADCA-II)
- ADCA mit rein zerebellarer Symptomatik (ADCA-III)
- ADCA mit Myoklonien und Taubheit (ADCA-IV)
- ADCA mit mentaler Retardierung (SCA13)
- Episodische Ataxie (EA) Typ 1–6
- Holmes-Syndrom
X-chromosomale Ataxien
Systematische Einteilung
Eine weitere Einteilung nach klinischen Kriterien:[11]
- Spinale Ataxien, spinale Heredoataxien
- Zerebellare Ataxien, zerebellare Heredoataxien
- Polyneuritisartige Ataxien
Weblinks
- S1-Leitlinie Ataxien der Deutschen Gesellschaft für Neurologie. In: AWMF online (Stand 2008)
- Europäisches Forschungsprojekt zu Spinocerebellären Ataxien (Seite auf Englisch)
- Deutsche Heredo-Ataxie Gesellschaft e.V.
Einzelnachweise
- S. Jarius, B. Wildemann: ‘Medusa head ataxia’: the expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 1: Anti-mGluR1, anti-Homer-3, anti-Sj/ITPR1 and anti-CARP VIII. In: J Neuroinflammation. Band 12, 2015, S. 166. (free)
- S. Jarius, B. Wildemann: ‘Medusa head ataxia’: the expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 2: Anti-PKC-gamma, anti-GluR-delta2, anti-Ca/ARHGAP26 and anti-VGCC. In: J Neuroinflammation. Band 12, 2015, S. 167. (free)
- S. Jarius, B. Wildemann: ‘Medusa head ataxia’: the expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 3: Anti-Yo/CDR2, anti-Nb/AP3B2, PCA-2, anti-Tr/DNER, other antibodies, diagnostic pitfalls, summary and outlook. In: J Neuroinflammation. Band 12, 2015, S. 168. (free)
- H. Mitoma, K. Adhikari, D. Aeschlimann, P. Chattopadhyay, M. Hadjivassiliou, C. S. Hampe u. a.: Consensus Paper: Neuroimmune Mechanisms of Cerebellar Ataxias. In: Cerebellum. Band 15, Nr. 2, 2016, S. 213–232, doi:10.1007/s12311-015-0664-x, PMID 25823827, PMC 4591117 (freier Volltext).
- A. Sapone, J. C. Bai, C. Ciacci, J. Dolinsek, P. H. Green, M. Hadjivassiliou, K. Kaukinen, K. Rostami, D. S. Sanders, M. Schumann, R. Ullrich, D. Villalta, U. Volta, C. Catassi, A. Fasano: Spectrum of gluten-related disorders: consensus on new nomenclature and classification. In: BMC Medicine. Band 10, 2012, S. 13, doi:10.1186/1741-7015-10-13, PMID 22313950, PMC 3292448 (freier Volltext).
- Ana Vinagre-Aragón, Panagiotis Zis, Richard Adam Grunewald, Marios Hadjivassiliou: Movement Disorders Related to Gluten Sensitivity: A Systematic Review. In: Nutrients. Band 10, Nr. 8, 8. August 2018, ISSN 2072-6643, doi:10.3390/nu10081034, PMID 30096784, PMC 6115931 (freier Volltext).
- Luis Rodrigo, Nuria Álvarez, Enrique Fernández-Bustillo, Javier Salas-Puig, Marcos Huerta: Efficacy of a Gluten-Free Diet in the Gilles de la Tourette Syndrome: A Pilot Study. In: Nutrients. Band 10, Nr. 5, 7. Mai 2018, ISSN 2072-6643, S. 573, doi:10.3390/nu10050573, PMID 29735930, PMC 5986453 (freier Volltext) – (mdpi.com [abgerufen am 14. Juni 2021]).
- M. Hadjivassiliou, D. D. Sanders, D. P. Aeschlimann: Gluten-related disorders: gluten ataxia. In: Dig Dis. Band 33, Nr. 2, 2015, S. 264–268, doi:10.1159/000369509, PMID 25925933.
- M. Hadjivassiliou, P. Aeschlimann, D. S. Sanders, M. Maki, K. Kaukinen: Transglutaminase 6 antibodies in the diagnosis of gluten ataxia. In: Neurology. Band 80, Nr. 19, 7. Mai 2013, ISSN 0028-3878, S. 1740–1745, doi:10.1212/WNL.0b013e3182919070 (neurology.org [abgerufen am 8. April 2020]).
- Vgl. auch Immo von Hattingberg: Spinale Heredoataxie (Friedreich) und Cerebellare Heredoataxie (Nonne, Pierre Marie). In: Ludwig Heilmeyer (Hrsg.): Lehrbuch der Inneren Medizin. Springer-Verlag, Berlin/ Göttingen/ Heidelberg 1955. (2. Auflage. ebenda 1961, S. 1347 f.)
- Flexikon