Chemische Reaktion
Eine chemische Reaktion ist ein Vorgang, bei dem eine oder meist mehrere chemische Verbindungen in andere umgewandelt werden und Energie freigesetzt oder aufgenommen wird. Auch Elemente können an Reaktionen beteiligt sein. Chemische Reaktionen sind in der Regel mit Veränderungen der chemischen Bindungen in Molekülen oder Kristallen verbunden. Durch eine chemische Reaktion können sich die Eigenschaften der Produkte im Vergleich zu den Edukten stark ändern. Nicht zu den chemischen Reaktionen zählen physikalische Vorgänge, bei denen sich lediglich der Aggregatzustand ändert wie Schmelzen oder Verdampfen, Diffusion, das Vermengen von Reinstoffen zu Stoffgemischen sowie Kernreaktionen, bei denen Elemente in andere umgewandelt werden.

Reaktionen bestehen aus einer meist recht komplizierten Folge einzelner Teilschritte, den sogenannten Elementarreaktionen, die zusammen die Gesamtreaktion bilden. Auskunft über den exakten Ablauf der Teilschritte gibt der Reaktionsmechanismus. Zur Beschreibung chemischer Reaktionen wird die Reaktionsgleichung verwendet, in der Edukte, Produkte und mitunter auch wichtige Zwischenprodukte graphisch dargestellt werden und über einen Pfeil, den Reaktionspfeil, miteinander verbunden werden.
Sowohl Elementarreaktionen als auch Reaktionsmechanismen kann man in verschiedene Gruppen aufteilen. Zu den Elementarreaktionen zählen etwa der Zerfall von einem Molekül in zwei oder der umgekehrte Fall, die Synthese von zwei Atomen oder Molekülen zu einem. Reaktionsmechanismen werden häufig nach der erfolgten Änderung in den beteiligten Stoffen eingeteilt. Erfolgt etwa eine Änderung der Oxidationszahlen, spricht man von Oxidation und Reduktion; entsteht ein festes Produkt aus gelösten Stoffen, von einer Fällung.
In welchem Umfang eine bestimmte Reaktion zweier oder mehrerer Partner stattfindet, hängt davon ab, wie groß die Differenz der sich aus einem enthalpischen und einem entropischen Anteil zusammensetzenden Gibbs-Energie der Produkte und der Edukte ist. Bei negativen Werten liegt das Reaktionsgleichgewicht auf Seite der Produkte. Es gibt jedoch auch viele Reaktionen, die zwar in diesem Sinne thermodynamisch möglich sind, aber kinetisch nur sehr langsam ablaufen, im Extremfall so langsam, dass sie praktisch nicht beobachtet werden können. Verantwortlich hierfür ist eine zu hohe Aktivierungsenergie, die aufgebracht werden muss, damit die weitere Reaktion möglich wird. Derartige Reaktionen laufen aber bei höheren Temperaturen schneller ab, da so eine vergleichsweise größere Anzahl der beteiligten Teilchen genug Energie besitzt um die Aktivierungsbarriere zu überwinden. Bei vielen Reaktionen ist dies auch mittels Katalyse möglich, bei der nicht die direkte Reaktion, sondern eine andere, bei der ein dritter, aus der Reaktion unverändert hervorgehender Stoff beteiligt ist, stattfindet. Durch die Anwesenheit dieses Katalysators wird die benötigte Aktivierungsenergie gesenkt.
Geschichte
.jpg.webp)
Chemische Reaktionen wie die Verbrennung im Feuer, die alkoholische Gärung oder die Reduktion von Erzen zu Metallen – beispielsweise bei Eisen – sind schon seit sehr langer Zeit bekannt. Erste Theorien zur Umwandlung von Stoffen wurden von griechischen Philosophen entwickelt, etwa die Vier-Elemente-Lehre des Empedokles, nach der jeder Stoff aus den vier Grundelementen Feuer, Wasser, Luft und Erde zusammengesetzt ist und in diese auch zerlegt werden kann. Im Mittelalter beschäftigten sich vor allem die Alchemisten mit chemischen Reaktionen. Dabei versuchten sie insbesondere Blei in Gold umzuwandeln, wobei sie unter anderem Reaktionen von Blei und Blei-Kupfer-Legierungen mit Schwefel einsetzten.[1]
Die Herstellung chemischer Substanzen, die in der Natur nicht vorkommen, durch geeignete Reaktionen ist schon lange bekannt. Dies betrifft etwa die Schwefel- und Salpetersäure, deren erstmalige Herstellung dem umstrittenen Alchemisten Dschābir ibn Hayyān zugeschrieben werden. Die Herstellung erfolgte durch Erhitzung von Sulfat- und Nitraterzen wie Kupfervitriol, Alaun und Salpeter. Im 17. Jahrhundert stellte Johann Rudolph Glauber durch Reaktion von Schwefelsäure und Natriumchlorid erstmals Salzsäure und Natriumsulfat her. Mit Entwicklung des Bleikammer-Verfahrens zur Schwefelsäureproduktion und des Leblanc-Verfahrens zur Natriumcarbonatherstellung wurden chemische Reaktionen auch industriell eingesetzt. Mit der zunehmenden Industrialisierung wurde die industrielle Synthese immer bedeutender und es wurden neuere und effizientere Verfahren entwickelt. Beispiele sind etwa das ab 1870 angewendete Kontaktverfahren zur Schwefelsäureproduktion oder das 1910 entwickelte Haber-Bosch-Verfahren zur Ammoniaksynthese.
Ab dem 16. Jahrhundert versuchten Forscher wie Johan Baptista van Helmont, Robert Boyle oder Isaac Newton beobachtete chemische Umwandlungen wissenschaftlich zu untersuchen und Theorien zu ihrem Ablauf aufzustellen. Eine wichtige untersuchte Reaktion war die Verbrennung, für die Johann Joachim Becher und Georg Ernst Stahl Anfang des 18. Jahrhunderts die Phlogistontheorie entwickelten. Diese erwies sich jedoch als falsch und konnte 1785 durch Antoine Lavoisier widerlegt werden, der die korrekte Erklärung der Verbrennung als Reaktion mit Sauerstoff der Luft fand.[2]
Joseph Louis Gay-Lussac erkannte 1808, dass Gase stets in bestimmten Verhältnissen miteinander reagieren. Daraus und aus Daltons Atomtheorie entwickelte Joseph Louis Proust das Gesetz der konstanten Proportionen, auf dem die Stöchiometrie aufbaut und das auch die Entwicklung der Reaktionsgleichungen ermöglichte.[3]
Für organische Reaktionen wurde lange Zeit angenommen, dass sie durch eine spezielle „Lebenskraft“ (vis vitalis) bestimmt werden und sich so von nicht-organischen Reaktionen unterscheiden. Nach der Harnstoffsynthese aus anorganischen Vorläufersubstanzen durch Friedrich Wöhler 1828 verlor diese Annahme in der Chemie stark an Bedeutung. Weitere Chemiker, die wichtige Beiträge zur Aufklärung organischer chemischer Reaktionen lieferten, waren beispielsweise Justus von Liebig mit seiner Radikaltheorie, Alexander William Williamson, der die nach ihm benannte Synthese von Ethern entwickelte, sowie Christopher Kelk Ingold, der unter anderem die Mechanismen für Substitutionsreaktionen erforschte.
Reaktionsgleichungen
Um chemische Reaktionen graphisch darzustellen, werden sogenannte Reaktionsgleichungen genutzt. Diese bestehen aus Summen- oder Strukturformeln der Edukte auf der linken und denen der Produkte auf der rechten Seite. Dazwischen befindet sich ein Pfeil, der sogenannte Reaktionspfeil, der die Richtung und Art der Reaktion anzeigt. Die Spitze des Pfeiles zeigt dabei immer in die Richtung, in die die Reaktion verläuft. Bei Gleichgewichtsreaktionen werden Doppelpfeile genutzt, die in entgegengesetzte Richtungen zeigen. Reaktionsgleichungen sollten stöchiometrisch ausgeglichen sein. Dies bedeutet, dass auf beiden Seiten des Reaktionspfeils die gleiche Zahl Atome stehen soll und Gleichungen gegebenenfalls durch unterschiedliche Anzahlen der beteiligten Moleküle ausgeglichen werden.[4]
- Schematische einfache Reaktionsgleichung

Kompliziertere Reaktionen werden durch Formelschemata dargestellt, die neben Edukten und Produkten auch wichtige Zwischenprodukte oder Übergangszustände zeigen. Dabei werden die Reaktionswege durch Pfeile verdeutlicht, die den Angriff von Elektronenpaaren eines Atoms an andere Atome zeigen. In Reaktionsgleichungen der organischen Chemie werden kleine, anorganische Moleküle wie Wasser oder Kohlenstoffdioxid, häufig auf den Pfeil (für Edukte) oder darunter (für Produkte) gesetzt oder durch Vorzeichen kenntlich gemacht. Auch Katalysatoren, Lösungsmittel, besondere Bedingungen oder andere Stoffe, die während der Reaktion eine Rolle spielen, sich bei dieser aber nicht verändern, werden auf den Reaktionspfeil geschrieben.
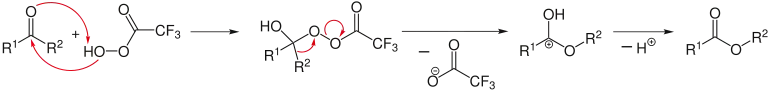 Typischer Reaktionsmechanismus der organischen Chemie (Beispiel: Baeyer-Villiger-Oxidation anhand der Reaktion einer Percarbonsäure mit einem Keton)
Typischer Reaktionsmechanismus der organischen Chemie (Beispiel: Baeyer-Villiger-Oxidation anhand der Reaktion einer Percarbonsäure mit einem Keton)
Für die Planung komplizierter Synthesen kann auch die Schreibweise einer Reaktion als Retrosynthese nützlich sein. Hier wird eine Reaktion vom Ende, also dem Produkt her aufgeschrieben, das über mögliche Syntheseschritte so lange zerlegt wird, bis mögliche Edukte erreicht sind. Retrosynthesen werden durch einen speziellen Pfeil, den Retrosynthesepfeil (), gekennzeichnet.[5]

Elementarreaktionen
Die Elementarreaktion ist der kleinste Abschnitt, in den eine chemische Reaktion zerlegt werden kann. Makroskopisch beobachtbare Reaktionen bauen sich aus einer Vielzahl Elementarreaktionen auf, die parallel oder nacheinander ablaufen. Die konkrete Abfolge einzelner Elementarreaktionen bezeichnet man auch als Reaktionsmechanismus. An einer Elementarreaktion sind in der Regel ein oder zwei, selten drei Moleküle beteiligt. Reaktionen mit mehr Molekülen sind praktisch ausgeschlossen, da es äußerst unwahrscheinlich ist, dass sich mehr als drei Moleküle gleichzeitig nahe genug für eine Reaktion kommen.[6]
Die wichtigsten Elementarreaktionen sind die unimolekularen und die bimolekularen Reaktionen. Bei einer unimolekularen Reaktion ist nur ein Molekül beteiligt, das sich durch eine Isomerisierung oder einen Zerfall in ein oder mehrere andere Moleküle umwandelt. Für diese Reaktionen braucht es in der Regel Energiezufuhr etwa in Form von Wärme oder durch Bestrahlung mit Licht.
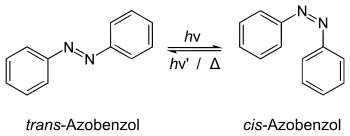
Ein Beispiel für eine typische unimolekulare Reaktion ist die cis-trans-Isomerisierung, bei der die cis-Form einer Verbindung in die trans-Form oder umgekehrt umgewandelt wird.
Bei einer Dissoziation spaltet sich eine Bindung in einem Molekül und es entstehen zwei Teile. Die Spaltung kann homo- oder heterolytisch erfolgen. Im ersten Fall wird die Bindung so gespalten, dass jeder Teil ein Elektron behält und Radikale entstehen, bei der heterolytischen Spaltung bleiben beide Elektronen bei einem Teil des Moleküls, während der andere keine Elektronen aus der gespaltenen Bindung zurückbehält und so Ionen entstehen. Zerfälle spielen eine wichtige Rolle beim Auslösen von Kettenreaktionen wie der Knallgasreaktion oder Polymerisationen.
- Zerfall eines Moleküls AB in zwei kleinere Teile A und B

Bei bimolekularen Reaktionen stoßen zwei Moleküle zusammen und reagieren miteinander. Eine Möglichkeit dabei ist, dass aus diesen zwei Molekülen ein einziges wird, also eine Synthese stattfindet. Dies erfolgt beispielsweise bei der Reaktion zweier Radikale zu einem Molekül. Auch bei Additionsreaktionen der organischen Chemie bildet sich aus mehreren Molekülen ein neues.

Es ist aber auch möglich, dass bei einer Reaktion kein stabiles Molekül entsteht und nur ein Teil des einen auf das andere Molekül übergeht. Dieser Reaktionstyp tritt beispielsweise bei Redox- und Säure-Base-Reaktionen auf. Bei Redoxreaktionen ist das übertragene Teilchen ein Elektron, bei Säure-Base-Reaktionen ein Proton. Dieser Reaktionstyp wird auch Metathese genannt.

Chemisches Gleichgewicht
Jede chemische Reaktion in homogener Phase ist umkehrbar und kann in beide Richtungen verlaufen. Wenn etwa die Reaktion zweier Stoffe zu einem dritten stattfindet, existiert gleichzeitig auch der Zerfall des dritten in die Ausgangsstoffe. Hin- und Rückreaktion stehen immer in Konkurrenz zueinander und unterscheiden sich durch unterschiedliche Reaktionsgeschwindigkeiten. Da Reaktionsgeschwindigkeiten auch konzentrationsabhängig sind, ändern sie sich mit der Zeit. Die Geschwindigkeiten von Hin- und Rückreaktion nähern sich mit Verlauf der Reaktion immer weiter an, bis sie schließlich gleich sind. Zu diesem Zeitpunkt ändern sich die Konzentrationen der einzelnen Stoffe in der Reaktionsmischung nicht mehr, ein Gleichgewicht, das sogenannte chemische Gleichgewicht, ist erreicht.
Die Lage des Gleichgewichtes ist neben den Eigenschaften der beteiligten Stoffe abhängig von der Temperatur und dem Druck und wird durch die minimale freie Energie bestimmt. Häufig wird auch mit der Ableitung der freien Enthalpie, der freien Reaktionsenthalpie gerechnet, die im Gleichgewicht 0 sein muss. Die Druckabhängigkeit lässt sich einfach mit dem Prinzip von Le Chatelier erklären, nach der ein System einem Zwang wie einer Druckerhöhung so ausweicht, dass die Wirkung minimal wird.
An diesem Punkt ist die maximale Ausbeute einer Reaktion erreicht, da bei weiterer Bildung eines Produktes nun die Rückreaktion schneller abläuft und daher so lange bevorzugt wird, bis wieder das Gleichgewicht erreicht wird. Größere Ausbeuten lassen sich aber durch Entfernen von Produkten aus der Reaktionsmischung, bei der das Gleichgewicht gestört wird, oder durch Änderungen von Druck oder Temperatur erzielen. Keinen Einfluss auf die Lage des Gleichgewichtes besitzen die Ausgangskonzentrationen der beteiligten Stoffe.
Thermodynamik
Chemische Reaktionen werden maßgeblich von den Gesetzen der Thermodynamik bestimmt. Prinzipiell läuft jede Reaktion ab. Jedoch liegt das Gleichgewicht in sehr vielen Fällen fast vollständig auf Seite der Edukte. Damit eine Reaktion ablaufen kann, muss sie exergon sein, also die freie Enthalpie während der Reaktion abnehmen. Die freie Enthalpie setzt sich aus zwei verschiedenen thermodynamischen Größen, der Enthalpie und der Entropie, zusammen. Diese sind über die Fundamentalgleichung für die freie Enthalpie miteinander verbunden.[7]
- G: freie Enthalpie, H: Enthalpie, T: Temperatur, S: Entropie, Δ: Differenzen

Reaktionen können auf mehrere Arten stattfinden. Eine Möglichkeit ist die exotherme Reaktion, bei der ΔH negativ ist und Energie frei wird. Abhängig von der Größe der freiwerdenden Energie können hierbei auch hochgeordnete Strukturen, die eine niedrige Entropie besitzen, entstehen. Typische Beispiele für exotherme Reaktionen mit Entropieverlust sind Fällungen und Kristallisationen, bei denen aus ungeordneten Strukturen in Gasphase, Flüssigkeit oder Lösung, geordnete feste Strukturen entstehen. Bei endothermen Reaktionen wird dagegen Wärme verbraucht und muss aus der Umgebung aufgenommen werden. Diese können nur ablaufen, wenn gleichzeitig die Entropie des Systems zunimmt. Dies kann beispielsweise über die Bildung gasförmiger Reaktionsprodukte erfolgen, die eine hohe Entropie besitzen.
Da die Entropie temperaturabhängig ist und mit steigender Temperatur zunimmt, finden Entropie-bestimmte Reaktionen wie Zerfälle bevorzugt bei hohen Temperaturen statt. Energie-bestimmte Reaktionen wie Kristallisationen finden dagegen vor allem bei tiefen Temperaturen statt. Mitunter lässt sich die Richtung einer Reaktion durch Temperaturänderung umdrehen.
Ein Beispiel hierfür ist das Boudouard-Gleichgewicht.

Die Reaktion von Kohlenstoffdioxid und Kohlenstoff zu Kohlenstoffmonoxid ist endotherm, so dass das Gleichgewicht bei tiefen Temperaturen auf der Seite des Kohlenstoffdioxides liegt. Erst bei Temperaturen von über 800 °C ist durch die höhere Entropie auf der Seite des Kohlenstoffmonoxides diese Seite bevorzugt.[8]
Auch über Änderungen der inneren Energie können Reaktionen betrachtet werden. Diese lässt sich ebenfalls über eine Fundamentalgleichung beschreiben, die unter anderem Entropie, Volumenänderungen und chemisches Potential berücksichtigt. Letzteres hängt unter anderem von den Aktivitäten der beteiligten Stoffe ab.[9]
- U: innere Energie, S: Entropie, p: Druck, μ: chemisches Potential, n: Stoffmenge, d: differentielle Schreibweise

Reaktionskinetik
Die Reaktionskinetik untersucht die Geschwindigkeit, mit der eine Reaktion abläuft. Diese ist von verschiedenen Parametern der Reaktion abhängig, etwa der Reaktionsordnung, den Konzentrationen der beteiligten Stoffe, der Temperatur, der Aktivierungsenergie und weiteren, meist empirisch bestimmten Faktoren. Zudem gibt es verschiedene Theorien, Reaktionsgeschwindigkeiten für verschiedene Systeme theoretisch auf molekularer Ebene zu berechnen. Dieses Arbeitsgebiet wird in Abgrenzung zur Reaktionskinetik auch als Reaktionsdynamik bezeichnet.
Für Elementarreaktionen lassen sich einfache Geschwindigkeitsgesetze aufstellen, die sich je nach Reaktionsordnung unterscheiden und die Abhängigkeit von den Konzentrationen der beteiligten Stoffe zeigen. Für eine Reaktion erster Ordnung, also etwa einen Zerfall eines Stoffes A, gilt für die Reaktionsgeschwindigkeit v (k: Geschwindigkeitskonstante, t: Zeit, [A]: Konzentration von A, [A]0: Anfangskonzentration von A):

integriert

Die Reaktionsgeschwindigkeit hängt also bei einer Reaktion erster Ordnung nur von der Konzentration und den Eigenschaften des zerfallenden Stoffes ab. Da bei einer Reaktion 1. Ordnung die Konzentration exponentiell mit der Zeit abnimmt, lässt sich eine konstante und damit für die jeweilige Reaktion typische Halbwertszeit bestimmen. Vor allem bei den nicht zu den chemischen Reaktionen gehörenden, aber ebenfalls nach einem Geschwindigkeitsgesetz erster Ordnung ablaufenden radioaktiven Zerfällen wird dieser Wert häufig angegeben. Für andere Reaktionsordnungen und kompliziertere Reaktionen existieren entsprechend andere Geschwindigkeitsgesetze. Um die Geschwindigkeitskonstante zu berechnen, kann die Arrhenius-Gleichung verwendet werden, die die Temperaturabhängigkeit der Konstante zeigt.
Ein einfaches Modell, mit dem der molekulare Ablauf einer chemischen Reaktion und die Reaktionsgeschwindigkeiten erklärt werden können, ist die Stoßtheorie. Mit dieser lassen sich jedoch nur wenige einfache Reaktionen einigermaßen korrekt berechnen. Für kompliziertere Reaktionen muss darum auf genauere Theorien, die in der Regel auf ein spezielles Problem zugeschnitten sind, zurückgegriffen werden. Dazu zählen etwa die Theorie des Übergangszustandes, die Berechnung der Potentialhyperfläche, die Marcus-Theorie und die RRKM-Theorie.[10]
Arten von Reaktionen
Reaktionen können in verschiedene Arten eingeteilt werden, die sich in der Art des übertragenen Teilchens und den entstehenden Produkten unterscheiden.
Oxidation und Reduktion
Werden bei einer Reaktion zweier Atome Elektronen übertragen, ändern sich die Oxidationsstufen der beteiligten Atome. Das Atom, das ein oder mehrere Elektronen abgibt (Reduktionsmittel genannt), wird oxidiert, das andere, das Oxidationsmittel, entsprechend reduziert. Da beide Reaktionen stets zusammen auftreten, spricht man auch von einer Redoxreaktion.
Welcher der beteiligten Reaktionspartner Reduktions- beziehungsweise Oxidationsmittel ist, lässt sich anhand der Elektronegativitäten der beteiligten Elemente vorhersagen. Elemente mit niedrigen Elektronegativitäten, wie die meisten Metalle, geben leicht Elektronen ab und werden dementsprechend oxidiert, Nichtmetalle mit hohen Elektronegativitäten werden dagegen leicht reduziert. Sind Ionen an einer Redoxreaktion beteiligt, ist auch die Oxidationsstufe des Ions zu beachten. So sind Chromate oder Permanganate, bei denen die Elemente in hohen Oxidationsstufen vorliegen, starke Oxidationsmittel.
Wie viele Elektronen ein Element in einer Redoxreaktion abgibt beziehungsweise aufnimmt, lässt sich häufig durch die Elektronenkonfiguration der Edukte vorhersagen. Elemente versuchen, die Edelgaskonfiguration zu erreichen und geben darum häufig eine dementsprechende Anzahl Elektronen ab oder nehmen sie auf. Dies gilt insbesondere für viele Hauptgruppenelemente wie die Alkalimetalle, Erdalkalimetalle oder Halogene. Für Übergangsmetalle und insbesondere schwere Atome gilt dies auf Grund der notwendigen hohen Ladung zum Erreichen der Edelgaskonfiguration und dem zunehmenden Einfluss relativistischer Effekte jedoch vielfach nicht. Die Edelgase, die schon Edelgaskonfiguration besitzen, haben dementsprechend keine Neigung, weitere Elektronen aufzunehmen und sind sehr reaktionsträge.
Eine wichtige Klasse der Redoxreaktionen sind die elektrochemischen Reaktionen. In der Elektrolyse dienen die Elektronen des elektrischen Stroms als Reduktionsmittel. Elektrochemische Reaktionen finden in galvanischen Zellen statt, bei denen Reduktion und Oxidation räumlich getrennt stattfinden. Besonders wichtig sind diese Reaktionen für die Gewinnung vieler Elemente wie Chlor oder Aluminium. Auch die umgekehrte Reaktion, bei der in Redoxreaktionen Elektronen frei werden und als elektrische Energie genutzt werden können, ist möglich. Dies ist das Prinzip der Batterie, in der Energie chemisch gespeichert und in elektrische Energie umgewandelt wird.
Komplexbildungsreaktion
Bei der Komplexbildungsreaktion reagieren mehrere Liganden mit einem Metallatom zu einem Komplex. Dies erfolgt dadurch, dass freie Elektronenpaare der Liganden in leere Orbitale des Metallatoms eindringen und eine koordinative Bindung bilden. Bei den Liganden handelt es sich um Lewis-Basen, die freie Elektronenpaare besitzen. Dies können sowohl Ionen als auch neutrale Moleküle (etwa Kohlenstoffmonoxid, Ammoniak oder Wasser) sein. In welcher Anzahl Liganden mit dem zentralen Metallatom reagieren, lässt sich häufig mit Hilfe der 18-Elektronen-Regel voraussagen, durch die besonders stabile Komplexe bestimmt werden können. Nach der Kristallfeld- und Ligandenfeldtheorie spielt auch die Geometrie des Komplexes eine wichtige Rolle, besonders häufig bilden sich tetraedrische oder oktaedrische Komplexe.[11]
Auch innerhalb eines Komplexes können Reaktionen stattfinden. Dazu zählen etwa der Ligandenaustausch, bei der ein oder mehrere Liganden durch einen anderen ersetzt werden, Umlagerungen sowie Redox-Vorgänge, bei denen sich die Oxidationsstufe des zentralen Metallatoms ändert.[11]
Säure-Base-Reaktionen
Säure-Base-Reaktionen sind – bei der Säuredefinition nach Brønsted – Reaktionen, bei denen Protonen von einem Molekül zum anderen übertragen werden. Dabei wird das Proton immer von der Säure (Protonendonator) auf die Base (Protonenakzeptor) übertragen (Protolyse).
- Säure-Base-Reaktion, HA: Säure, B: Base, A−: korrespondierende Base, HB+: korrespondierende Säure

Da bei der Übertragung des Protons von der Säure zur Base wiederum eine Base und eine Säure entstehen, die sogenannten korrespondierenden Säuren bzw. Basen, ist auch die Rückreaktion möglich. Säure/Base und korrespondierende Base/Säure stehen daher immer im Gleichgewicht. Auf welcher Seite der Reaktion das Gleichgewicht liegt, lässt sich durch die Säurekonstanten der beteiligten Stoffe bestimmen. Je stärker eine Säure bzw. Base ist, desto leichter gibt sie das Proton ab bzw. nimmt es auf. Ein Spezialfall der Säure-Base-Reaktion ist die Neutralisation, bei der eine Säure und eine Base in exakt dem Verhältnis reagieren, dass eine neutrale Lösung, also eine Lösung ohne Überschuss an Hydroxid- oder Oxoniumionen entsteht.
Fällung
Die Fällung ist eine Reaktion, bei der sich vorher gelöste Teilchen verbinden und zu einem neuen, wasserunlöslichen Stoff werden, dem Niederschlag. Dies findet vor allem bei gelösten Ionen statt, die sich bei Überschreitung des Löslichkeitsproduktes zusammenfinden und ein unlösliches Salz bilden. Dies kann beispielsweise durch Zugabe eines Fällungsmittels mit geringem Löslichkeitsprodukt zu einem schon gelösten Salz oder durch Entfernen des Lösungsmittels erfolgen. Je nach Bedingungen kann ein Stoff sehr unterschiedlich aus einer Lösung ausfallen. Erfolgt die Fällung schnell, haben die Ionen keine Zeit sich zu ordnen, es bildet sich ein amorpher oder mikrokristalliner Niederschlag. Beim langsamen Überschreiten des Löslichkeitsproduktes und einer Übersättigung erfolgt die Fällung dagegen nur langsam. Die Ionen haben daher Zeit, sich zu ordnen und es bilden sich regelmäßig aufgebaute Kristalle. Dies kann auch durch Umkristallisation aus dem mikrokristallinen Niederschlag erfolgen.[12]
Festkörperreaktionen
Reaktionen können auch zwischen zwei festen Stoffen stattfinden. Jedoch ist die Diffusion, die die Reaktionsgeschwindigkeit hierbei maßgeblich bestimmt, sehr klein, Festkörperreaktionen sind dementsprechend langsame Reaktionen. Dies bewirkt, dass Festkörperreaktionen in der Regel bei hohen Temperaturen durchgeführt werden müssen. Gleichzeitig sollten die Reaktanten möglichst fein verteilt vorliegen, da so eine möglichst große Oberfläche, an der die beiden Stoffe reagieren können, geschaffen wird.[13]
Photochemische Reaktionen
In photochemischen Reaktionen spielt elektromagnetische Strahlung eine entscheidende Rolle. Von besonderer Bedeutung sind hierbei Licht und UV-Strahlung von etwa 200 bis 800 nm Wellenlänge. Durch diese Strahlung werden Elektronen in Atomen und Molekülen angeregt, es bilden sich angeregte Zustände. Diese sind durch die absorbierten Photonen sehr energiereich und können die Energie über verschiedene Prozesse abgeben. Neben physikalischen Prozessen wie Fluoreszenz und Phosphoreszenz sind hier auch Reaktionen möglich. Häufig erfolgen homolytische Bindungsbrüche, so dass Radikale entstehen. So können durch photochemische Reaktionen beispielsweise Kettenreaktionen wie die Knallgasreaktion von Wasserstoff und Sauerstoff ausgelöst werden. Aber auch Ionisierungen, Elektronentransferreaktionen, Isomerisierungen oder Umlagerungen können durch photochemische Reaktionen verursacht werden.[14]
Eine biologisch sehr wichtige photochemische Reaktion ist die Photosynthese, bei der mit Hilfe von Licht organische Verbindungen aus Kohlenstoffdioxid und Wasser synthetisiert werden. Auch in der Atmosphärenchemie, etwa beim Auf- und Abbau von Ozon spielen photochemische Reaktionen eine wichtige Rolle.
Katalyse
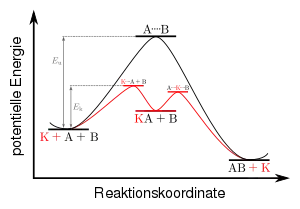
Bei einer Katalyse findet die Reaktion zweier Stoffe nicht direkt, sondern über einen Umweg statt. Es ist immer ein dritter Stoff, der sogenannte Katalysator, beteiligt, der in die Reaktion eingreift, aber am Ende stets unverändert aus der Reaktion hervorgeht. Durch die Katalyse können Reaktionen, die durch eine hohe Aktivierungsenergie kinetisch gehemmt wird, unter Umgehung dieser Aktivierungsenergie stattfinden. Dadurch ist häufig nur noch ein geringer Energieeinsatz und damit eine wirtschaftliche Durchführung einer Reaktion möglich. Mitunter werden Reaktionen durch Katalysatoren auch erst ermöglicht, wenn etwa bei sonst nötigen Temperaturen Konkurrenzreaktionen bevorzugt ablaufen.
Katalysatoren können sowohl in einer anderen Phase (heterogen) als auch in gleicher Phase (homogen) vorliegen. Heterogene Katalysatoren sind meist Festkörper, an deren Oberfläche die Reaktionen stattfinden. Dementsprechend sollte die Oberfläche des Katalysators für eine effektive Katalyse möglichst groß sein. Katalytische Reaktionen an Oberflächen sind häufig mit Chemisorption verbunden, bei der ein Molekül chemisch an die Oberfläche gebunden und daher die Bindungen innerhalb des Moleküls geschwächt werden. So ist eine leichtere Reaktion möglich.
Von besonderer Bedeutung in der heterogenen Katalyse sind die Platinmetalle und weitere Übergangsmetalle, die in vielen technisch wichtigen Reaktionen wie Hydrierungen, Katalytisches Reforming oder der Synthese von Grundchemikalien wie Salpetersäure oder Ammoniak verwendet werden. Katalysatoren der Homogenen Katalyse können etwa Säuren sein, die die Nukleophilie einer Carbonylgruppe erhöhen und so eine Reaktion mit sonst nicht reagierenden Elektrophilen ermöglichen, oder lösliche Komplexe wie bei der Hydroformylierung.
Homogene Katalysatoren haben den Vorteil, dass es keine Probleme mit der Erreichbarkeit des Katalysators und zu kleinen Oberflächen gibt, die Reaktanten und der Katalysator können durch Vermischen und Rühren leicht zusammengebracht werden. Zudem kann der Katalysator, etwa ein Komplex, speziell und reproduzierbar für eine Reaktion synthetisiert werden. Ein Nachteil ist jedoch die schwierige Abtrennung des Katalysators vom Produkt, was zu Verunreinigungen und Verlust des meist teuren Katalysators führen kann. Darum werden in vielen technischen Prozessen heterogene Katalysatoren bevorzugt.[15]
Reaktionen in der organischen Chemie
In der organischen Chemie gibt es neben den auch bei anorganischen Stoffen ablaufenden Reaktionen wie Oxidationen, Reduktionen oder Säure-Base-Reaktionen eine Vielzahl weiterer Reaktionen, bei denen kovalente Bindungen zwischen Kohlenstoffatomen oder Kohlenstoff- und Heteroatomen (beispielsweise Sauerstoff, Stickstoff, Halogene) gebildet werden. Diese werden neben der Unterscheidung zwischen homolytischen und radikalisch ablaufenden Reaktionen, vor allem nach der Art der Strukturänderung eingeteilt. Viele spezielle Reaktionen in der organischen Chemie sind als Namensreaktionen nach ihren Entdeckern benannt.
Substitution
Bei der Substitution wird ein Atom, Molekülteil oder Ligand (in der Komplexchemie, in der Substitutionen ebenfalls möglich sind) gegen einen anderen ausgetauscht. Ein angreifendes Atom oder Molekül nimmt dabei den Platz eines anderen, als Abgangsgruppe abgespaltenen Atoms oder Moleküls ein. Die Bindigkeit des Kohlenstoffatoms ändert sich nicht.
Substitutionsreaktionen können je nach angreifendem Teilchen in drei Arten eingeteilt werden. Bei nukleophilen Substitutionen greift ein Nukleophil, also ein Atom oder Molekül mit einem Elektronenüberschuss und damit einer negativen Ladung oder Partialladung, an ein geeignetes Kohlenstoffatom an und ersetzt ein anderes Atom oder Teilmolekül. Typische Nukleophile sind Atome, Ionen oder Atomgruppen mit elektronegativen Nichtmetallen wie Amine, Halogenide, Thiole, Hydroxide oder Alkoholate. Neben dem Nukleophil spielt auch die Abgangsgruppe eine Rolle, ob eine Substitution stattfindet. Gute Abgangsgruppen sollten leicht abzuspalten sein und möglichst stabile Moleküle oder Ionen bilden. Beispiele sind die schweren Halogenide Bromid und Iodid oder Stickstoff. Diesen Reaktionstyp findet man vorwiegend bei aliphatischen Kohlenwasserstoffen, bei Aromaten ist er – da Aromaten eine hohe Elektronendichte besitzen – eher selten und kann nur unter speziellen Umständen bei sehr stark elektronenziehenden Gruppen am Aromaten stattfinden (Nukleophile aromatische Substitution). Nukleophile Substitutionen können nach zwei verschiedenen Mechanismen, als SN1 und SN2 bezeichnet, ablaufen. Die Bezeichnungen leiten sich von den Reaktionsordnungen ab, nach denen die geschwindigkeitsbestimmenden Schritte der beiden Reaktionsarten ablaufen.
Im SN1-Mechanismus wird zunächst die Abgangsgruppe abgespalten, es entsteht ein Carbokation. Anschließend erfolgt eine schnelle Reaktion mit dem Nukleophil.
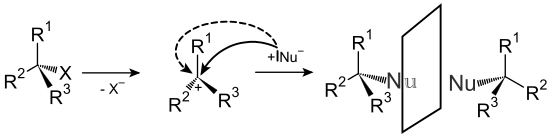
Beim SN2-Mechanismus greift zunächst das Nukleophil unter Bildung eines gemeinsamen Übergangszustandes an, erst danach wird die Abgangsgruppe abgespalten. Die beiden Mechanismen unterscheiden sich in der Stereochemie der erhaltenen Produkte, bei SN1 tritt auf Grund des dreibindigen Carbokations eine Racemisierung ein, während bei SN2 eine Umkehr der vorher vorhandenen Stereochemie (Walden-Umkehr) beobachtet wird.[16]
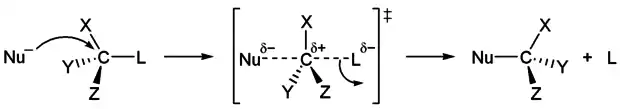
Das Gegenstück zur nukleophilen Substitution ist die Elektrophile Substitution. Bei dieser ist ein Elektrophil, also ein Atom oder Molekül mit einer geringeren Elektronendichte, also einer positiven Ladung oder Partialladung, das angreifende Teilchen. Typische Elektrophile sind beispielsweise Carbokationen, das Kohlenstoffatom in Carbonylgruppen, Schwefeltrioxid oder Nitronium-Kationen. Diese Reaktion findet fast ausschließlich bei aromatischen Kohlenwasserstoffen statt, man spricht darum auch häufig von einer elektrophilen aromatischen Substitution. Im Mechanismus bildet sich durch den Angriff des Elektrophils zunächst der sogenannte σ-Komplex, ein Übergangszustand, bei dem das aromatische System aufgehoben ist. Anschließend wird die Abgangsgruppe, in der Regel ein Proton, abgespalten und das aromatische System wiederhergestellt.[17]

Bei der dritten Substitutionsart ist das angreifende Teilchen ein Radikal, es wird darum auch von einer radikalischen Substitution gesprochen. Diese verläuft in Form einer Kettenreaktion und findet beispielsweise bei der Reaktion von Alkanen mit Halogenen statt. Im ersten Schritt werden etwa durch Licht, Hitze oder den Zerfall von sehr instabilen Molekülen wenige Startradikale gebildet. In der Kettenreaktion verläuft die Reaktion durch Übertragung des Radikals weiter, bis es durch die Rekombination zweier Radikale zu einem Kettenabbruch kommt.[18]
- Reaktionen während der Kettenreaktion einer radikalischen Substitution


Addition/Eliminierung
Die Addition und das Gegenstück, die Eliminierung, sind Reaktionen, bei denen sich die Anzahl der Substituenten am Kohlenstoffatom ändert und Mehrfachbindungen gebildet oder gespalten werden. Bei Eliminierungsreaktionen werden Doppel- und Dreifachbindungen aufgebaut, indem an jedem Kohlenstoffatom der Bindung jeweils ein Substituent entfernt („eliminiert“) wird. Für eine Eliminierung muss sich an einem Kohlenstoffatom der fraglichen Bindung eine geeignete Abgangsgruppe befinden, die relativ leicht abgespalten werden kann. Ähnlich wie bei der nukleophilen Substitution gibt es mehrere mögliche Mechanismen, die je nach Molekül und Bedingungen ablaufen und wiederum nach der jeweiligen Reaktionsordnung benannt sind. Im E1-Mechanismus findet zunächst die Abspaltung der Abgangsgruppe unter Bildung eines Carbokations statt. Im nächsten Schritt erfolgt dann die Ausbildung der Doppelbindung unter Abspaltung eines Protons. Auf Grund der ähnlichen Bedingungen beider Reaktionen steht die E1-Eliminierung immer in Konkurrenz zur SN1-Substitution.[19]

Ebenfalls erster Reaktionsordnung ist der E1cb-Mechanismus, bei dem mit Hilfe einer Base zunächst das Proton abgespalten wird und sich ein Carbanion bildet. Im nächsten Schritt bildet sich unter Abspaltung der Abgangsgruppe die Doppelbindung.[20]

Der E2-Mechanismus erfordert ebenfalls eine Base. Bei diesem laufen jedoch der Angriff der Base und die Abspaltung der Abgangsgruppe konzertiert ab und es wird kein ionisches Zwischenprodukt gebildet. Im Gegensatz zu den E1-Eliminierungen ist hier die Festlegung der Stereochemie im Produkt möglich, da eine Reaktion der Base in anti-Stellung zur Abgangsgruppe bevorzugt abläuft. Durch ähnliche Bedingungen und Reagenzien steht die E2-Eliminierung immer in Konkurrenz zur SN2-Substitution.[21]

Das Gegenstück zur Eliminierung ist die Additionsreaktion. Bei dieser lagern sich Atome oder Moleküle an Doppel- oder Dreifachbindungen an und bilden Einfachbindungen. Additionsreaktionen können sowohl an C-C-Mehrfachbindungen, also Alkenen oder Alkinen, als auch an Kohlenstoff-Heteroatom-Mehrfachbindungen wie Carbonylgruppen, Thiocarbonylgruppen oder Nitrilen stattfinden. Wie die Substitutionen lassen sich auch die Additionen je nach angreifendem Teilchen in mehrere Gruppen einteilen. Bei der elektrophilen Addition greift ein Elektrophil, häufig ein Proton, an der Doppelbindung unter Bildung eines Carbeniumions an. Dieses reagiert mit Nukleophilen unter Bildung des Produktes.
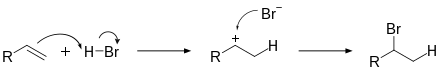
Für die Bildung des Carbeniumions gibt es zwei Möglichkeiten – auf welcher Seite der Doppelbindung es bevorzugt gebildet wird, hängt bei asymmetrischen Alkenen von der Stabilisierung durch unterschiedliche Reste ab. Eine Regel, welches der Produkte bevorzugt gebildet wird, bietet die Markownikow-Regel.
Soll die Addition einer funktionellen Gruppe am weniger substituierten Kohlenstoffatom der Doppelbindung stattfinden, ist die elektrophile Substitution mit Säuren nicht möglich. Eine Möglichkeit bietet die Hydroborierung, bei der das Boratom als Elektrophil wirkt und daher entsprechend der Markownikow-Regel am weniger substituierten Kohlenstoffatom angreift. Durch Oxidation oder Halogenierung können in einem weiteren Schritt dann andere funktionelle Gruppen gebildet werden.[22]
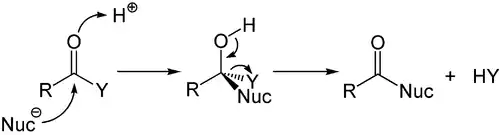
Während bei den elektronenreichen Alkenen und Alkinen vor allem die elektrophile Addition auftritt, spielt bei den Kohlenstoff-Heteroatom-Mehrfachbindungen und vor allem deren wichtigstem Vertreter, der Carbonylgruppe, die nukleophile Addition eine wichtige Rolle. Diese ist häufig mit einer Eliminierung verbunden, so dass nach der Reaktion die Carbonylgruppe wieder vorliegt. Dieses kann bei Carbonsäurederivaten wie Carbonsäurechloriden, -estern oder -anhydriden erfolgen, die eine geeignete Abgangsgruppe an der Carbonylgruppe tragen. Es wird dabei häufig vom Additions-Eliminierungs-Mechanismus gesprochen. Dieser wird häufig durch Säuren oder Basen katalysiert, die (bei Säuren) durch Anlagerung an das Sauerstoffatom die Elektrophilie der Carbonylgruppe oder (bei Basen) die Nukleophilie des angreifenden Nukleophils erhöhen.[23]
Ein Angriff durch eine nukleophile Addition kann gemäß dem Vinylogie-Prinzip auch an die Doppelbindung von α,β-ungesättigten Carbonylverbindungen wie Ketonen oder Estern stattfinden. Ein wichtiger Vertreter dieser Reaktionsart ist die Michael-Addition.[24]
Additionen können wie Substitutionen nicht nur durch Nukleophile und Elektrophile, sondern auch durch Radikale ausgelöst werden. Wie bei der radikalischen Substitution verläuft auch die radikalische Addition in Form einer Kettenreaktion. Diese Reaktion ist die Grundlage der radikalischen Polymerisation.[25]
Weitere organische Reaktionsmechanismen
Umlagerungen sind Reaktionen, bei denen die Atome oder Molekülteile einer organischen Verbindung erhalten bleiben, aber neu angeordnet werden. Hierzu zählen Hydridverschiebungs-Reaktionen wie die Wagner-Meerwein-Umlagerung, bei der zunächst ein Carbokation gebildet wird, das sich anschließend unter Verschiebung eines Hydrid-Iones zu einem stabileren Carbokation umlagert. Meist sind Umlagerungen jedoch mit dem Brechen und Neubilden von C-C-Bindungen verbunden. Typische Beispiele hierfür sind sigmatrope Umlagerungen wie die Cope-Umlagerung, bei der in einer cyclischen Reaktion gleichzeitig eine C-C-Bindung gebrochen und eine andere gebildet wird.[26]
Wie die sigmatropen Umlagerungen gehören auch Cycloadditionen zu den pericyclischen Reaktionen. Bei dieser Reaktion wird aus mehreren, meist zwei, Doppelbindungen enthaltenden Molekülen ein cyclisches Molekül gebildet. Die wichtigste Cycloaddition ist die Diels-Alder-Reaktion, eine [4+2]-Cycloaddition, bei der ein Dien und ein Alken (auch als Dienophil bezeichnet) zu einem Cycloalken reagieren.
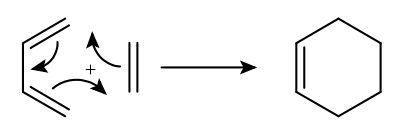
Neben der Diels-Alder-Reaktion gibt es auch die [2+2]-Cycloaddition, bei der zwei Alkene oder andere Verbindungen mit Doppelbindungen wie Ketone miteinander reagieren. Mit 1,3-Dipolen wie Ozon, Diazomethan oder Nitriloxiden sind ebenfalls Cycloadditionen möglich. Ob und wie eine Cycloaddition abläuft, hängt von der Anordnung der p-Orbitale der beteiligten Doppelbindungen ab.
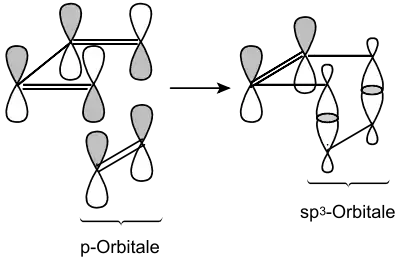
Diese müssen so gegeneinanderstehen, dass jeweils Orbitale mit dem gleichen Vorzeichen der Wellenfunktion überlappen und damit konstruktiv wechselwirken können und die energetisch günstigeren Einfachbindungen bilden. Cycloreaktionen können sowohl thermisch als auch photochemisch durch Bestrahlung mit Licht induziert werden. Da bei der Bestrahlung Elektronen in Orbitale gebracht werden, die eine andere Anordnung und Symmetrie besitzen, sind photochemisch andere Cycloadditionen möglich als thermisch. So sind Diels-Alder-Reaktionen thermische Cycloadditionen, während [2+2]-Cycloadditionen durch Bestrahlungen induziert werden müssen.[27]
Durch die Orbitalanordnungen werden die möglichen entstehenden Produkte und -bei stereoisomeren Edukten- auch deren Stereoisomerie eingeschränkt. Wie dies stattfindet, wird durch die Woodward-Hoffmann-Regeln beschrieben.[28]
Biochemische Reaktionen
In biochemischen Reaktionen sind Enzyme von zentraler Bedeutung. Diese Proteine katalysieren meist speziell eine einzelne Reaktion, so dass Reaktionen sehr exakt gesteuert werden können. Es sind aber auch Enzyme bekannt, die mehrere spezielle Funktionen katalytisch beschleunigen können. Die Reaktion findet in einem kleinen Teil des Enzyms, dem Aktiven Zentrum statt, während der Rest des Enzyms überwiegend zur Stabilisierung dient. Das aktive Zentrum liegt in einer Grube oder Furche des Enzyms. Für die katalytische Aktivität sind unter anderem Bindungen an das Enzym, die veränderte, hydrophobe, chemische Umgebung und die räumliche Nähe der Reaktanten verantwortlich, während die spezielle Form des aktiven Zentrums für die Selektivität verantwortlich ist.[29]
Die Gesamtheit der biochemischen Reaktionen im Körper bezeichnet man als Stoffwechsel. Zu den wichtigsten Mechanismen zählt der Baustoffwechsel, bei dem in unterschiedlichen durch die DNA und Enzyme gesteuerten Prozessen wie der Proteinbiosynthese aus einfachen Vorläufersubstanzen komplexe Naturstoffe wie Proteine oder Kohlenhydrate synthetisiert werden. Daneben existiert der Energiestoffwechsel, durch den mit Hilfe chemischer Reaktionen die für eine Reaktion, etwa des Baustoffwechsels, notwendige Energie bereitgestellt wird. Ein wichtiger Energielieferant ist die Glucose, die durch Pflanzen in der Photosynthese hergestellt werden kann oder mit der Nahrung aufgenommen wird. Diese ist jedoch nicht direkt nutzbar, stattdessen wird über die Zellatmung und die Atmungskette mit Hilfe von Sauerstoff ATP erzeugt, das als Energielieferant für die weiteren Reaktionen dient.
Technische Anwendung
Chemische Reaktionen und ihre Durchführung sind zentral für die technische Chemie. Sie werden in großer Zahl zur Synthese neuer Verbindungen aus natürlich vorkommenden Grundstoffen wie Erdöl, Erzen, Luft oder nachwachsenden Rohstoffen eingesetzt. Häufig werden zunächst einfache Zwischenprodukte synthetisiert, aus denen dann die Endprodukte wie Polymere, Waschmittel, Pflanzenschutzmittel, Pharmaka oder Farbstoffe hergestellt werden. Technische Reaktionen finden in Reaktoren wie Rührkesseln oder Strömungsrohren statt.
Für die Technik ist es besonders wichtig, die Reaktionsführung so wirtschaftlich wie möglich zu gestalten. Dazu zählen etwa ein minimaler Rohstoff- und Energieeinsatz, hohe Reaktionsgeschwindigkeiten und hohe Ausbeuten mit möglichst wenigen Abfallprodukten. Von großer Bedeutung ist daher der Einsatz von Katalysatoren, die sowohl die Reaktionsgeschwindigkeit erhöhen als auch den Energieeinsatz verringern.[30] Um geringe Abfallmengen zu gewährleisten, werden in technischen Anwendungen häufig Reaktionen gewählt, die eine hohe Atomökonomie aufweisen, bei denen also ein Großteil der Edukte sich im gewünschten Produkt wiederfindet.[31]
Beobachtung
Wie chemische Reaktionen beobachtet und verfolgt werden können, hängt stark von der Reaktionsgeschwindigkeit ab. Bei langsamen Reaktionen können während der Reaktion Proben entnommen und analysiert werden. Dabei werden die Konzentrationen der einzelnen Inhaltsstoffe der Reaktionsmischung bestimmt und so der Konzentrationsverlauf während der Reaktion verfolgt. Ändern sich die Konzentrationen nach einiger Zeit nicht mehr, ist die Reaktion abgeschlossen und im Gleichgewicht angekommen. Damit die Reaktion während der Messung nicht zu sehr voranschreitet, werden vor allem schnelle und einfach durchzuführende Analyseverfahren wie die Dünnschichtchromatographie oder Massenspektrometrie eingesetzt. Auch eine kontinuierliche Beobachtung während der Reaktion ist durch spektroskopische Methoden möglich, wenn damit beispielsweise die Konzentration einer farbigen Substanz in der Mischung bestimmt werden kann. Ist dies nicht möglich, kann mitunter auch ein spezieller Marker, etwa ein radioaktives Isotop eingesetzt werden, dessen Konzentration dann gemessen wird. Dies wird beispielsweise in der Szintigrafie für die Beobachtung von Stoffwechselvorgängen eingesetzt, bei denen sich bestimmte Elemente in einzelnen Organen anreichern. Oberflächenreaktionen können unter günstigen Voraussetzungen direkt mit einem Rastertunnelmikroskop auf molekularer Ebene beobachtet werden.[32]
Bei Nachweisreaktionen spielen sogenannte Indikatoren eine wichtige Rolle, das sind Stoffe, die sich beispielsweise in ihrer Farbe verändern, wenn ein bestimmter Punkt der Reaktion erreicht ist. Bekannt sind vor allem Säure-Base-Indikatoren, die ihre Farbe ändern, sobald eine Lösung neutralisiert wurde und der pH-Wert vom Sauren ins Basische wechselt oder umgekehrt. Auch selektive Fällungsreaktionen können zum Nachweis von Stoffen oder etwa im Kationentrennungsgang zur Auftrennung vor dem genauen Nachweis genutzt werden.
Je schneller eine Reaktion abläuft, desto schwieriger wird es, sie zu beobachten. Für kinetische Untersuchungen schneller Reaktionen wird die Ultrakurzzeit-Spektroskopie verwendet, die mit Hilfe von Femtosekundenlasern eine Zeitauflösung im Bereich von Piko- oder Femtosekunden ermöglicht. So lassen sich auch kurzlebige Übergangszustände während der Reaktion beobachten.[33]
Literatur
- Eintrag zu Reaktion. In: Römpp Online. Georg Thieme Verlag, abgerufen am 20. Juni 2014.
- Peter W. Atkins, Julio de Paula: Physikalische Chemie. 4. Auflage, Wiley-VCH, Weinheim 2006, ISBN 978-3-527-31546-8.
- A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 102. Auflage. Walter de Gruyter, Berlin 2007, ISBN 978-3-11-017770-1, S. 186–258.
- Reinhard Brückner: Reaktionsmechanismen. 3. Auflage, Spektrum Akademischer Verlag, München 2004, ISBN 3-8274-1579-9.
Weblinks
- Eintrag zu chemical reaction. In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.C01033.
Einzelnachweise
- Jost Weyer: Neuere Interpretationsmöglichkeiten der Alchemie. In: Chemie in unserer Zeit. 1973, 7,6, S. 177–181, doi:10.1002/ciuz.19730070604.
- William H. Brock: Viewegs Geschichte der Chemie. Vieweg, Braunschweig 1997, ISBN 3-540-67033-5, S. 34–55.
- William H. Brock: Viewegs Geschichte der Chemie. Vieweg, Braunschweig 1997, ISBN 3-540-67033-5, S. 104–107.
- Eintrag zu chemical reaction equation. In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.C01034.
- E. J. Corey: Robert Robinson Lecture. Retrosynthetic thinking—essentials and examples. In: Chem. Soc. Rev. 1988, 17, S. 111–133, doi:10.1039/CS9881700111.
- Eintrag zu Elementarreaktionen. In: Römpp Online. Georg Thieme Verlag, abgerufen am 20. Juni 2014.
- Peter W. Atkins, Julio de Paula: Physikalische Chemie. 4. Auflage, Wiley-VCH, Weinheim 2006, ISBN 978-3-527-31546-8, S. 106–108.
- A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 102. Auflage. Walter de Gruyter, Berlin 2007, ISBN 978-3-11-017770-1, S. 897.
- Peter W. Atkins, Julio de Paula: Physikalische Chemie. 4. Auflage, Wiley-VCH, Weinheim 2006, ISBN 978-3-527-31546-8, S. 150.
- Peter W. Atkins, Julio de Paula: Physikalische Chemie. 4. Auflage, Wiley-VCH, Weinheim 2006, ISBN 978-3-527-31546-8, S. 963.
- A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 102. Auflage. Walter de Gruyter, Berlin 2007, ISBN 978-3-11-017770-1, S. 1380–1400.
- Eintrag zu Ausfällung. In: Römpp Online. Georg Thieme Verlag, abgerufen am 20. Juni 2014.
- Ralf Alsfasser, Erwin Riedel, C. Janiak, H. J. Meyer: Moderne anorganische Chemie. 3. Auflage. de Gruyter, 2007, ISBN 978-3-11-019060-1, S. 171.
- Peter W. Atkins, Julio de Paula: Physikalische Chemie. 4. Auflage, Wiley-VCH, Weinheim 2006, ISBN 978-3-527-31546-8, S. 937–950.
- Christoph Elschenbroich: Organometallchemie. 6. Auflage, Teubner Wiesbaden, 2008, ISBN 978-3-8351-0167-8, S. 263.
- Reinhard Brückner: Reaktionsmechanismen. 3. Auflage, Spektrum Akademischer Verlag, München 2004, ISBN 3-8274-1579-9, S. 63–77.
- Reinhard Brückner: Reaktionsmechanismen. 3. Auflage, Spektrum Akademischer Verlag, München 2004, ISBN 3-8274-1579-9, S. 203–206.
- Reinhard Brückner: Reaktionsmechanismen. 3. Auflage, Spektrum Akademischer Verlag, München 2004, ISBN 3-8274-1579-9, S. 16.
- Reinhard Brückner: Reaktionsmechanismen. 3. Auflage, Spektrum Akademischer Verlag, München 2004, ISBN 3-8274-1579-9, S. 183.
- Reinhard Brückner: Reaktionsmechanismen. 3. Auflage, Spektrum Akademischer Verlag, München 2004, ISBN 3-8274-1579-9, S. 192.
- Reinhard Brückner: Reaktionsmechanismen. 3. Auflage, Spektrum Akademischer Verlag, München 2004, ISBN 3-8274-1579-9, S. 172.
- Reinhard Brückner: Reaktionsmechanismen. 3. Auflage, Spektrum Akademischer Verlag, München 2004, ISBN 3-8274-1579-9, S. 125.
- Hans Peter Latscha, Uli Kazmaier, Helmut Alfons Klein: Organische Chemie: Chemie-basiswissen II, Band 2. 6. Auflage, Springer, 2008, ISBN 978-3-540-77106-7, S. 273.
- Reinhard Brückner: Reaktionsmechanismen. 3. Auflage, Spektrum Akademischer Verlag, München 2004, ISBN 3-8274-1579-9, S. 580.
- Manfred Lechner, Klaus Gehrke, Eckhard Nordmeier: Makromolekulare Chemie. 3. Auflage, Birkhäuser, Basel 2003, ISBN 3-7643-6952-3, S. 53–65.
- Eberhard Breitmaier, Günther Jung: Organische Chemie. 5. Auflage, Thieme, Stuttgart 2005, ISBN 3-13-541505-8, S. 447–453.
- Reinhard Brückner: Reaktionsmechanismen. 3. Auflage, Spektrum Akademischer Verlag, München 2004, ISBN 3-8274-1579-9, S. 637–647.
- R. B. Woodward, Roald Hoffmann: Stereochemistry of Electrocyclic Reactions. In: J. Am. Chem. Soc. 1965, 87, 2, S. 395–397, doi:10.1021/ja01080a054.
- Peter Karlson, Detlef Doenecke, Jan Koolman, Georg Fuchs, Wolfgang Gerok: Karlsons Biochemie und Pathobiochemie. 16. Auflage, Georg Thieme Verlag, 2005, ISBN 978-3-13-357815-8, S. 55–56.
- Gerhard Emig, Elias Klemm: Technische Chemie. 5. Auflage, Springer, 2005, ISBN 978-3-540-23452-4, S. 33–34.
- Barry Trost: The atom economy-a search for synthetic efficiency. In: Science. 1991, 254, S. 1471–1477, doi:10.1126/science.1962206.
- Thomas Waldmann, Daniela Künzel, Harry E. Hoster, Axel Groß, R. Jürgen Behm: Oxidation of an Organic Adlayer: A Bird’s Eye View. In: Journal of the American Chemical Society. 134, Nr. 21, 2012, S. 8817–8822, doi:10.1021/ja302593v.
- Peter W. Atkins, Julio de Paula: Physikalische Chemie. 4. Auflage, Wiley-VCH, Weinheim 2006, ISBN 978-3-527-31546-8, S. 987.