Konjugation (Chemie)
Unter Konjugation versteht man in der Chemie die abwechselnde Überlappung einer π-Bindung (π = Pi) mit verschiedenen σ-Bindungen zwischen jeweils zwei sp2-hybridisierten (Kohlenstoff-)Atomen oder mit weiteren π-Bindungen. Im ersten Fall (dies entspricht konjugierten Radikalen, Carbokationen und Carbanionen) besteht die Atomkette aus einer ungeraden Anzahl an Atomen bzw. p-Orbitalen, bei konjugierten Doppelbindungen hingegen aus einer geraden Anzahl an Atomen bzw. p-Orbitalen. Die aus der Überlappung resultierenden Molekülorbitale ergeben sich aus dem Konzept der MO-Theorie. Konjugation führt zu π-Systemen mit delokalisierten Elektronen. Eng verwandt ist der Begriff der Mesomerie. Bei cyclischen, planaren, konjugierten Systemen kann Aromatizität auftreten.
Auswirkungen auf Reaktivität und Struktur
Radikale, Carbokationen und Carbanionen
Alle drei Spezies werden durch Konjugation stabilisiert. Der Grund ist, dass durch die Delokalisation der Elektronen über mehrere Atome hinweg sich der Bereich ihres möglichen Aufenthalts vergrößert. Da gemäß dem Modell Teilchen im Kasten die Energie eines Teilchens umgekehrt proportional zum Quadrat der Kastenausdehnung ist, ist auch hier die Energie der Teilchen geringer. Konjugierte Radikale, Carbokationen und Carbanionen sind durch diesen Effekt thermodynamisch stabiler als nicht-konjugierte. Diese Stabilisierung bezeichnet man als Konjugationsenergie. Gemäß dem Bell-Evans-Polanyi-Prinzip entstehen konjugierte Zwischenstufen in Reaktionen im Vergleich somit schneller.
Da für eine Überlappung die p-Orbitale parallel ausgerichtet sein müssen, befinden sich alle Substituenten an den sp2-hybridisierten Atomen in einer Ebene.
Polyene
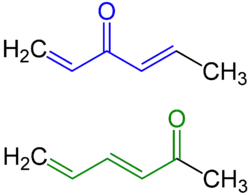
Konjugierte Polyene, d. h. Polyene, in denen die einzelnen Doppelbindungen nur durch eine C–C-Einfachbindung getrennt sind, profitieren auch von der Konjugationsenergie. Da sie dementsprechend thermodynamisch stabiler als ihre nicht-konjugierte Analoga sind, verlaufen analoge Reaktionen, wie die Hydrierung, die Addition von H–Hal, Hal2, H2O, o. ä. oder die Reaktion mit Hydroperoxiden zu Epoxiden – wiederum gemäß Bell-Evans-Polanyi – langsamer.
Die Bindung zwischen zwei durch eine Einfachbindung getrennten Kohlenstoffatomen in einem konjugierten System ist mit 148 pm kürzer als eine normale C–C-Einfachbindung (154 pm). Dies liegt an zwei Effekten: Als sp2-hybridisierte Kohlenstoffatome sind die Bindungspartner elektronegativer als sp3-hybridisierte Kohlenstoffatome. Sie ziehen das bindende Elektronenpaar stärker an, was eine Verkürzung bewirkt. Der zweite ist die Konjugation: die π-Orbitale können überlappen. Dies ist der sog. partielle Doppelbindungscharakter. Er äußert sich auch in einer erhöhten Rotationsbarriere um diese Einfachbindung (dieser Effekt tritt auch in z. B. der C–N-Einfachbindung in Amiden auf).
Bei konjugierten Dienen ist es, im Zusammenhang mit den Woodward-Fieser-Regeln zur Berechnung des UV-Absorptionsmaximums wichtig, zu unterscheiden, ob die beiden Doppelbindungen Teil eines Ringes sind (homoannular) oder sich auf zwei Ringe verteilen (heteroannular).[1]
Allyl- und Benzylhalogenide
Allyl- oder benzylderivatsubstituierte Abgangsgruppen reagieren mit Nukleophilen nach dem SN2-Mechanismus schneller, da es im Übergangszustand zu Orbitalwechselwirkungen kommt, die die Ladung durch Delokalisation stabilisieren.
Betrachtungen mit der VB-Theorie
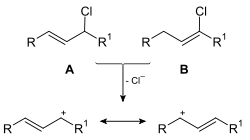 Abb. 1: Heterolyse zweier substituierter Propenderivate zum identischen Carbokation
Abb. 1: Heterolyse zweier substituierter Propenderivate zum identischen Carbokation
Gemäß den Regeln der Mesomerielehre lässt sich die Ladung bzw. das einzelne Elektron auf mehrere Atome verteilen (delokalisieren). Die formulierbaren Strukturen sind Grenzformeln ein und derselben Verbindung. So ergibt die Heterolyse der C–Cl-Bindung der Verbindungen A und B in Abb. 1 identische Kationen. Da allgemein die Verteilung von Ladung oder Elektronenmangelzentren energetisch begünstigt ist, lässt sich so die erhöhte Stabilität der konjugierten Spezies erklären.
Im Rahmen der Valenzstrukturtheorie (Valence Bond oder VB-Theorie) wird die Wellenfunktion als Linearkombination von chemisch interpretierbaren Strukturen geschrieben und bildet somit eine quantenmechanische Beschreibung des Mesomeriekonzeptes.[2][3] Mittels moderner quantenchemischer VB-Programme lassen sich auch die Gewichte der verschiedenen Resonanzstrukturen berechnen (d. h. welchen Anteil eine Resonanzstruktur an der Gesamtwellenfunktion hat) oder die Eigenenergien der hypothetischen einzelnen Mesomeristrukturen.[4][5] Durch das Konzept der Resonanz lassen sich auch die Reaktivität und Struktureigenschaften von konjugierten Doppelbindungen mit der VB-Theorie erklären und verstehen.[6] Für eine vollständige Beschreibung der Wellenfunktion können – je nach elektronischer Struktur der Moleküle – auch zwitterionische oder biradikalische[7] Strukturen eine wichtige Rolle spielen.[3] Zur Beschreibung der chemischen Reaktivität können ferner sogenannte Valence Bond Diagramme herangezogen werden.[8]
Betrachtungen mit der MO-Theorie
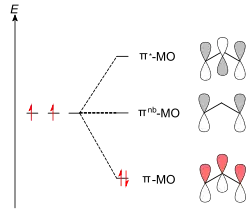
In der MO-Theorie werden n Atomorbitale zu n Molekülorbitalen kombiniert (LCAO-Methode). Bei einer ungeraden Anzahl an zu kombinierenden p-AOs ergeben sich (n-1)/2 bindende, (n-1)/2 antibindende und ein nichtbindendes MO. Dies wird hier am Beispiel des Propenylkations demonstriert (Abb. 2). Alle drei p-AOs besitzen hier dieselbe Energie. Diese dürfen auf drei unterschiedliche Arten kombiniert werden, so dass drei Molekülorbitale resultieren. Die Elektronen populieren nach dem Besetzungsprinzip das MO mit der niedrigsten Energie.
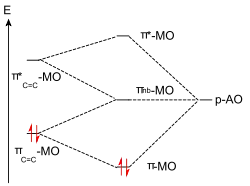
Eine im Ergebnis gleiche Alternative ist zwei Atomorbitale zu einem π- und einem π*-Orbital zu kombinieren und diese beiden mit dem verbliebenen p-Atomorbital zu den drei Molekülorbitalen zu kombinieren (Abb. 3). In dieser Vorgehensweise lässt sich die Konjugationsenergie erkennen, die das Molekül im Vergleich zu einer isolierten Doppelbindung und einem isolierten Elektronensextett am Kohlenstoff stabilisiert: sie beträgt den doppelten (da zwei Elektronen) energetischen Abstand des πC=C-Orbitals zum π-MO des Moleküls.
Siehe auch
Einzelnachweise
- Joseph B. Lambert, Scott Gronert, Herbert F. Shurvell, David A. Lightner: Spektroskopie – Strukturaufklärung in der Organischen Chemie. 2. Auflage. Pearson Deutschland, München 2012, ISBN 978-3-86894-146-3, S. 646–653.
- S. Shaik, P. C. Hiberty: Valence Bond theory, its History, Fundamentals and Applications. A Primer. In: Reviews of Computational Chemistry. Band 20, 2004, Kapitel 1.
- Sason S. Shaik, Philippe C. Hiberty: A Chemist's Guide to Valence Bond Theory. John Wiley & Sons, 2007, ISBN 978-0-470-19258-0.
- L. Song, Y. Mo, Q. Zhang, W. Wu: XMVB: A program for ab initio nonorthogonal valence bond computations. In: Journal of Computational Chemistry. Band 26, Nr. 5, 2005, S. 514.
- J. Li, R. McWeeny: VB2000: Pushing Valence Bond Theory to new limits. In: International Journal of Quantum Chemistry. Band 89, Nr. 4, 2002, S. 208.
- In älteren Quellen findet sich noch manchmal die Aussage, dass die VB-Theorie diese und andere Konzepte nicht erklären kann. Jedoch basiert diese Aussage auf einem falschen Verständnis der Theorie. Das Kapitel 5 (Are the "Failures" of Valence Bond Theorie Real?) des Buches "A Chemist's Guide to Valence Bond Theory" von Shaik ud Hiberty setzt sich mit diesem Sachverhalt auseinander.
- Benoit Braida, Christof Walter, Bernd Engels, Philippe C. Hiberty: A Clear Correlation between the Diradical Character of 1,3-Dipoles and Their Reactivity toward Ethylene or Acetylene. In: Journal of the American Chemical Society. Band 132, Nr. 22, 9. Juni 2010, S. 7631–7637, doi:10.1021/ja100512d.
- Sason Shaik, David Danovich, Hui Chen, Chunsen Li, Wenzhen Lai: A tutorial for understanding chemical reactivity through the valence bond approach. In: Chemical Society Reviews. Band 43, Nr. 14, 23. Juni 2014, ISSN 1460-4744, S. 4968–4988, doi:10.1039/C4CS00043A (rsc.org [abgerufen am 17. August 2019]).