Chinolin
Chinolin, auch Azanaphthalin oder Benzo[b]pyridin genannt, ist eine organische Verbindung aus der Gruppe der Heteroaromaten und gehört zu den zweikernigen heterocyclischen Stammsystemen. Sie besteht aus zwei anellierten aromatischen sechsgliedrigen Ringen – einem Benzol- und einem Pyridinring –, woraus sich die Summenformel C9H7N ergibt. Formal handelt es sich somit um ein Naphthalinmolekül, bei welchem ein Kohlenstoffatom des Ringgerüsts durch ein Stickstoffatom ausgetauscht wurde. Chinolin ist eine farblose, wasserbindende Flüssigkeit mit unangenehmem, stechendem Geruch. Als Heteroaromat weist Chinolin bezüglich der elektrophilen aromatischen Substitution eine geringere Reaktivität als Naphthalin auf, geht jedoch im Vergleich leichter nukleophile aromatische Substitutionen ein.
| Strukturformel | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
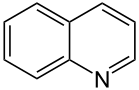 | ||||||||||||||||
| Allgemeines | ||||||||||||||||
| Name | Chinolin | |||||||||||||||
| Andere Namen |
| |||||||||||||||
| Summenformel | C9H7N | |||||||||||||||
| Kurzbeschreibung |
farblose bis gelbliche, stark lichtbrechende, hygroskopische Flüssigkeit[1], farblose, zerfließliche Prismen als Hydrochlorid·Hydrat[2] | |||||||||||||||
| Externe Identifikatoren/Datenbanken | ||||||||||||||||
| ||||||||||||||||
| Eigenschaften | ||||||||||||||||
| Molare Masse | 129,16 g·mol−1 | |||||||||||||||
| Aggregatzustand |
flüssig | |||||||||||||||
| Dichte |
1,10 g·cm−3 (20 °C)[3] | |||||||||||||||
| Schmelzpunkt |
| |||||||||||||||
| Siedepunkt |
237,2 °C[3] | |||||||||||||||
| Dampfdruck | ||||||||||||||||
| pKS-Wert |
4,94 (konjugierte Säure)[5] | |||||||||||||||
| Löslichkeit |
| |||||||||||||||
| Dipolmoment | ||||||||||||||||
| Brechungsindex |
1,6262 (21 °C)[7] | |||||||||||||||
| Sicherheitshinweise | ||||||||||||||||
| ||||||||||||||||
| Toxikologische Daten | ||||||||||||||||
| Thermodynamische Eigenschaften | ||||||||||||||||
| ΔHf0 |
141,22 kJ·mol−1[10] | |||||||||||||||
| Soweit möglich und gebräuchlich, werden SI-Einheiten verwendet. Wenn nicht anders vermerkt, gelten die angegebenen Daten bei Standardbedingungen. Brechungsindex: Na-D-Linie, 20 °C | ||||||||||||||||



Chinolin ist in Steinkohlenteer enthalten und kann aus Kohle ausgetrieben werden. Es existieren zahlreiche natürliche Derivate des Chinolins, welche oftmals als Alkaloide in Pflanzen anzutreffen sind (siehe Chinolin-Alkaloide). Zu dieser Gruppe gehören die China-Alkaloide mit Chinin als bekanntestem Vertreter. Chinolin wurde erstmals 1834 von Friedlieb Ferdinand Runge aus Steinkohlenteer in reiner Form isoliert. 1842 wurde es von Charles Frédéric Gerhardt durch den Abbau des Alkaloids Cinchonin erhalten, von welchem sich auch die Bezeichnung Chinolin ableitet.
Chinolin ist ein bedeutender Grundstoff in der chemischen und pharmazeutischen Industrie. Es wird zur Herstellung von Arzneimitteln, Herbiziden und Fungiziden sowie als basisches Lösungsmittel eingesetzt. Obwohl mehrere synthetische Zugänge zu Chinolin bekannt sind, wird aus ökonomischen Gründen bis heute noch ein Großteil des weltweiten Chinolinbedarfs durch Isolation aus Steinkohlenteer gedeckt.
Geschichte

Die Isolierung reinen Chinolins gelang erstmals Friedlieb Ferdinand Runge im Jahre 1834.[11] Dieser extrahierte es aus Steinkohlenteer und gab der Verbindung den Namen Leucolin. 1842 wurde Chinolin ein zweites Mal von Charles Frédéric Gerhardt entdeckt, der die Zersetzungsprodukte von Chinin[12] und Cinchonin[13] durch Einwirkung von Alkalien analysierte und vermeintlich in beiden Fällen die gleiche, seiner Kenntnis nach bis dato unbekannte chemische Verbindung erhielt.
„Folgende Thatsache ist nicht uninteressant. Das Chinin verwandelt sich, unter dem Einflusse von Aetzkali, in eine neue stickstoffhaltige, bei gewöhnlicher Temperatur ölartige Base. Diese neue Verbindung, welche ich Chinolin nenne…“
Die Namensgebung erfolgte in Anlehnung an die Verbindungen Chinin und Cinchonin, aus welchen er Chinolin gewonnen hatte. 1843 bezeichnete Gerhardt die Verbindung als Chinoleïn,[13] später wurde auch Quinolein (vergleiche auch englisch Quinoline) verwendet.[14] Gerhardt irrte jedoch in der Annahme, dass Chinolin als Abbauprodukt sowohl von Chinin als auch von Cinchonin auftritt, denn wie später gezeigt wurde entsteht durch Zersetzung von Chinin ein methoxyliertes Chinolinderivat, wohingegen nur der Abbau von Cinchonin unsubstituiertes Chinolin liefert.[15][16] Die molekularen Strukturen von Runges Leucolin und Gerhardts Chinolin war zum Zeitpunkt der Entdeckung noch unbekannt. Erst 1882 wurde die Identität der beiden Verbindungen durch Hoogewerff und van Dorp eindeutig geklärt.[17]
« Les expériences décrites dans le pages précédentes semplent démontret qu’il y a dans le goudron de houille une leucoline identique à la quinoléine obtenue de la cinchonine. »
„Die auf den vorigen Seiten beschriebenen Versuchsergebnisse scheinen darauf hinzudeuten, dass im Steinkohlenteer ein Leucolin existiert, welches mit dem aus Cinchonin erhaltenen Chinolin identisch ist.“
Ab diesem Zeitpunkt begann sich die Bezeichnung Chinolin durchzusetzen.[15]
Die Aufklärung der Molekularstruktur des Chinolins gelang im Jahre 1879. Zuvor wurde bereits postuliert, bei Chinolin handele es sich um Naphthalin, bei welchem ein Kohlenstoffatom des Rings durch Stickstoff ersetzt sei. Da Naphthalin einige Jahre zuvor durch Cyclisierung von 4-Phenyl-1-buten dargestellt werden konnte,[18] könnte diese Hypothese durch die analoge Cyclisierung von N-Allylanilin zu Chinolin bestätigt werden. Dies gelang Koenigs im Jahre 1880 unter Verwendung von Blei(II)-oxid,[19] wodurch die postulierte Struktur von Chinolin als Naphthalin-Analogon erhärtet werden konnte.
In den folgenden Jahrzehnten wurden in zahlreichen Untersuchungen die chemischen und physikalischen Eigenschaften der Verbindung geklärt und verschiedene Synthesewege für Chinolin und dessen Derivate etabliert.
Vorkommen

Chinolin kommt in der Natur praktisch nicht frei vor, bildet jedoch das Grundgerüst zahlreicher Naturstoffe, aus denen es durch Abbaureaktionen freigesetzt werden kann. Hierzu gehört die Gruppe der China-Alkaloide, welche in hoher Konzentration in Chinarindenbäumen auftreten.[20] Chinolin findet sich zu etwa 0,3 % – zusammen mit vielen weiteren heterocyclischen Verbindungen – im Steinkohlenteer, einem Nebenprodukt der Koksgewinnung aus Steinkohle.[11] In Tabakrauch ist Chinolin analytisch nachweisbar.[21]
Nomenklatur
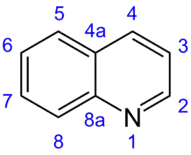
Neben Chinolin sind auch die Bezeichnungen Benzopyridin und 1-Azanaphthalin gelegentlich anzutreffen.[4] Benzopyridin beschreibt das Molekül als Pyridin mit anelliertem Benzolring und ist ohne weitere Qualifikatoren nicht eindeutig, da diese Bezeichnung auch auf das isomere Isochinolin zutrifft; korrekt und eindeutig ist Benzo[b]pyridin. 1-Azanaphthalin beschreibt Chinolin als Derivat von Naphthalin, bei welchem das Kohlenstoffatom (oder besser die Methingruppe) in 1-Position durch ein Stickstoffatom ausgetauscht wurde. Im Gegensatz zu Benzopyridin ist diese Bezeichnung eindeutig.
Die Nummerierung der Ringatome folgt der allgemeinen Regel für mehrkernige aromatische Systeme. Hierbei wird die Zählung am höchstrangigen Stammsystem begonnen, welches im vorliegenden Fall der Pyridinring ist. Dem Stickstoffatom wird hierbei als Heteroatom die höchste Priorität und damit eine möglichst kleine Nummer zugewiesen. Im Falle des Chinolins besitzt das Stickstoffatom die Nummer 1 und die Kohlenstoffringatome des Pyridinrings werden von dort aus fortlaufend mit 2–4 durchnummeriert. Die Zählweise wird fortlaufend im Benzolring weitergeführt, wobei die Brückenatome übersprungen werden. Diesen werden nach dem allgemeinen Nummerierungsschema für kondensierte Ringsysteme die Bezeichnungen 4a und 8a zugewiesen.[22]
Die systematische Bezeichnung des Chinolinrestes lautet Chinolyl, wobei die Position der Verknüpfung als Zahl vorangestellt wird. Analog dem Pyridinrest (Pyridyl anstatt systematisch Pyridinyl) wird die systematische Bezeichnung Chinolinyl nur selten verwendet. Die Verschmelzungskomponente von Chinolin als Stammsystem in kondensierten polycyclischen aromatischen Systemen lautet Chino.[22]
Gewinnung und Darstellung
Chinolin kann ebenso wie eine Reihe weiterer heterocyclischer Stickstoffbasen (beispielsweise Pyridin[23] und Pyrrol[24]) aus Steinkohlenteer gewonnen werden, in welchem es zu 0,3 % enthalten ist. Im Gegensatz zu Pyridin, dessen weltweiter Bedarf heutzutage im Wesentlichen durch synthetische Verfahren gedeckt wird,[25] erfolgt die Gewinnung von Chinolin auch heute noch zu großen Teilen aus Steinkohlenteer. Durch fraktionierte Destillation geht es zusammen mit Isochinolin und Chinaldin in der Methylnaphthalin-Fraktion über, aus welcher es mit Schwefelsäure zusammen mit Methylnaphthalin und Isochinolin extrahiert wird. Die anschließende Abtrennung vom Methylnaphthalin erfolgt durch Ausfällung mit Ammoniak. Auf Grund des um 6 °C höheren Siedepunkts von Isochinolin kann das verbleibende Gemisch aus Chinolin und Isochinolin durch Rektifikation aufgetrennt werden. Zur weiteren Aufreinigung von Chinolin bestehen verschiedene Möglichkeiten, wie die Verharzung von Verunreinigungen mit Formaldehyd, die Behandlung mit Alkalien, die selektive Oxidation sowie die Bildung von Hydraten (Isochinolin bildet keine Hydrate).[11] Des Weiteren kann Chinolin auch durch Azeotroprektifikation mit Ethylenglycol oder Diethylenglycol direkt aus der Methylnaphthalin-Fraktion erhalten und anschließend destillativ gereinigt werden.[26][27]
Skraup-Synthese
Nach Koenigs erster Chinolinsynthese[19] aus dem Jahre 1879 stellt die Skraup-Synthese, welche von Zdenko Hans Skraup im Jahre 1880 erstmals publiziert wurde[28] den zweiten synthetischen Zugang zu Chinolin dar. Sie geht aus von Anilin, welches in Gegenwart von Glycerin, Schwefelsäure und einem Oxidationsmittel zu Chinolin umgesetzt wird. Glycerin wird hierbei zunächst zu Acrolein dehydratisiert, welches als α,β-ungesättigte Carbonylverbindung zur Cyclisierung benötigt wird. Die direkte Verwendung von Acrolein verringert hingegen die Ausbeute, da es unter den Reaktionsbedingungen zur Polymerisation neigt. Die Skraup-Synthese liefert zunächst Dihydrochinolin, das durch milde Oxidationsmittel wie dreiwertige Eisensalze, Nitrobenzol oder Iod zu Chinolin oxidiert werden kann.[29] Die Skraup-Synthese stellt einen der wenigen direkten Synthesewege zu unsubstituiertem Chinolin dar.[5]
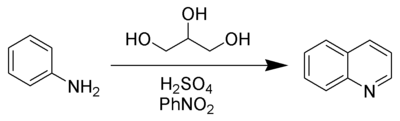
Die Doebner-Miller-Reaktion ist eine Abwandlung der Skraup-Synthese und setzt direkt α,β-ungesättigte Aldehyde ein, wodurch in 2-Position substituierte Chinoline hergestellt werden können.[30][31]
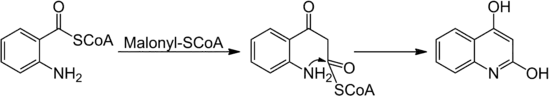
Friedländer-Chinolin-Synthese
Die Friedländer-Chinolin-Synthese geht aus von o-Aminobenzaldehyd, welches mit enolisierbaren Carbonylverbindungen zu Derivaten von Chinolin cyclisiert wird. Die Reaktion wird durch Trifluoressigsäure,[32] Toluolsulfonsäure,[33] Iod[34] und verschiedene Lewis-Säuren[35] katalysiert.
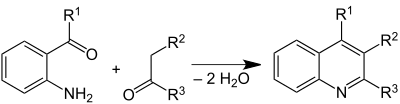
Die Niemantowski-Synthese ist eine Abwandlung der Friedländer-Chinolinsynthese, welche anstelle von o-Aminobenzaldehyden von Anthranilsäure ausgeht.[36]
Weitere Syntheserouten
Es sind eine Reihe weiterer Syntheserouten zu Chinolin beziehungsweise dessen Derivaten bekannt.[37] Namentlich seien die Conrad-Limpach-Synthese (nach Max Conrad, Leonhard Limpach), bei welcher Aniline und β-Ketoester eingesetzt werden,[38] und die Povarov-Reaktion, zu der Anilin, Benzaldehyd und aktivierte Alkene benötigt werden,[31] erwähnt. Ferner finden die Camps-Chinolinsynthese,[39] die Knorr-Chinolinsynthese[40] und die Gould-Jacobs-Reaktion[41] Anwendung.
Einige Chinolinalkaloide treten als Naturstoffe in biologischen Systemen auf. Der exakte biosynthetische Aufbau des Chinolins ist abhängig vom biologischen System und der genauen Struktur des Chinolinderivats. Den biochemischen Zugang einiger Chinolinderivate stellt die Aminosäure Tryptophan dar, aus welcher in einer mehrstufigen Reaktion das Chinolingerüst aufgebaut werden kann. Über einen weiteren Syntheseweg ausgehend von Anthranilsäure sind hydroxylierte Chinolingerüste zugänglich. Die Fortführung der Synthese führt zu Acridinderivaten.[42]
Eigenschaften
Physikalische Eigenschaften
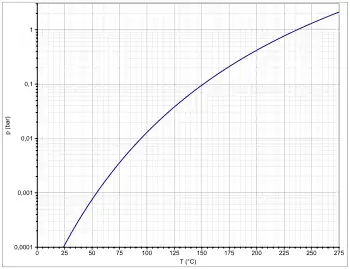
Chinolin ist farblos und bei Standardbedingungen flüssig.[1] Es siedet bei 237,2 °C und gefriert bei −14,8 °C.[3] Die Dampfdruckfunktion ergibt sich nach Antoine entsprechend log10(P) = A−(B/(T+C)) (P in bar, T in K) mit A = 3,94043, B = 1667,104 und C = −87,085 im Temperaturbereich von 437,8 bis 511,1 K.[43] Es ist eine stark lichtbrechende Flüssigkeit, die bei 21 °C und einer Wellenlänge von 589 nm einen Brechungsindex von 1,6262 aufweist.[7] Bei Standardbedingungen besitzt Chinolin eine mit Wasser vergleichbare Dichte von 1,10 g·cm−3.[3] Chinolin ist diamagnetisch (die molare diamagnetische Suszeptibilität beträgt −86,1·10−6 cm³·mol−1)[44] und weist ein Dipolmoment von 2,29 D auf.[6] Die kritische Temperatur beträgt 527 °C, der kritische Druck 57,8 bar.[45] in der flüssigen Phase beträgt die Standardbildungsenthalpie 141,22 kJ·mol−1,[10] in der Gasphase hingegen 200,5 kJ·mol−1.[46] Bei 25 °C besitzt Chinolin eine Viskosität von 3,337 mPa·s−1[47] und eine Wärmeleitfähigkeit von 0,147 W·(m·K)−1.[48]
Als Festkörper tritt die Verbindung in zwei polymorphen Kristallformen auf. Die Umwandlung von Kristallform II in Kristallform I erfolgt bei −53 °C.[10] Die Kristallform II kristallisiert im monoklinen Kristallsystem in der Raumgruppe P21/c (Raumgruppen-Nr. 14) mit den bei 150 K (−123 °C) bestimmten Gitterparametern a = 992 pm, b = 1085 pm, c = 1337 pm und β = 106,5° sowie acht Formeleinheiten je Elementarzelle. Die einzelnen Moleküle sind dabei in zwei orthogonal zueinander stehenden Ketten angeordnet, die über schwache C-H-N-Wasserstoffbrücken zusammengehalten werden. Zwischen den Ketten bestehen Wechselwirkungen zwischen C–H-Bindungen und dem aromatischen π-System.[49]
In Wasser ist Chinolin nur wenig löslich. So lösen sich bei 20 °C lediglich 6 g je Liter.[9] Seine deutlich schlechtere Löslichkeit im Vergleich zu Pyridin, das mit Wasser frei mischbar ist[50], ist dem unpolaren Benzolring geschuldet. Hingegen ist Chinolin mit Ethanol, Diethylether, Aceton, Benzol und Kohlenstoffdisulfid frei mischbar.[3]
Chemische Eigenschaften
Chinolin reagiert schwach basisch und bildet in Gegenwart von Salzsäure ein kristallines Hydrochlorid (C9H7N · HCl), welches bei 134 °C schmilzt.[2]
Chinolin gehört zur Gruppe der heteroaromatischen Verbindungen, deren Eigenschaften sich in seiner Reaktivität widerspiegeln. Im Vergleich zu seinem Kohlenstoffanalogon Naphthalin ist es weniger reaktionsfreudig bezüglich der elektrophilen Substitutionen, was auf die elektronenziehenden Eigenschaften des Stickstoffatoms zurückzuführen ist, welches zum einen die Elektronendichte im aromatischen System herabsetzt und zum anderen mit angreifenden Elektrophilen unter Bildung von nochmals elektronenärmeren Chinoliniumverbindungen zu reagieren vermag. Im Gegensatz zu Naphthalin besitzt Chinolin jedoch eine vergleichsweise höhere Reaktivität bezüglich nukleophiler Substitutionen. Das Stickstoffatom ist sp2-hybridisiert und weist typische basische Eigenschaften eines Amins auf. Elektrophile aromatische Substitutionen finden bevorzugt am Benzolring statt, während nukleophile aromatische Substitutionen eher am Pyridinring ablaufen.[29]
Analog dem Pyridin führt die Reaktion mit vielen Lewis-Säuren zur Addition an das Stickstoffatom.
Molekulare Eigenschaften
Chinolin weist ein durchkonjugiertes System mit zehn π-Elektronen auf, welche über das gesamte Ringsystem delokalisiert sind. Das heteroaromatische Molekül ist planar gebaut, aber die Elektronendichte ist nicht gleichmäßig verteilt, was auf den negativen induktiven Effekt des Stickstoffatoms zurückzuführen ist. Aus diesem Grund weist Chinolin analog dem Pyridin ein Dipolmoment auf.[6]
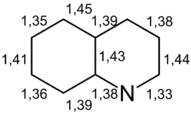
Die Bindungen im Molekül weisen unterschiedliche Längen auf. Als Ligand im Nickelkomplex betragen sie zwischen 133 und 145 pm und liegen somit wie für aromatische Systeme üblich zwischen den Werten, welche typischerweise für einzelgebundene und doppeltgebundene Atome erwartet werden. Naphthalin weist im Vergleich hierzu C–C-Bindungslängen zwischen 135 und 142 pm auf,[51] was auf eine gleichmäßigere Elektronenverteilung in diesem Molekül schließen lässt.
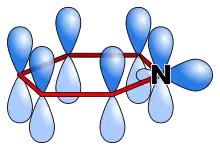
Im Chinolinmolekül sind alle Ringatome sp2-hybridisiert. Das Stickstoffatom stellt das Elektron seines p-Orbitals zur Ausbildung des aromatischen Systems zur Verfügung, sein freies sp2-Elektronenpaar liegt in der Molekülebene und weist vom Ringzentrum fort. Auf Grund seiner Position kann es nicht mit dem π-System in Wechselwirkung treten und trägt somit nicht zur Ausbildung der Aromatizität bei. Es ist jedoch bedeutend für die chemischen Eigenschaften von Chinolin, denn im Gegensatz zu Naphthalin wird das aromatische System durch Anlagerung eines Elektrophils an dieser Position nicht aufgehoben. Die Trennung des freien Elektronenpaars vom aromatischen System bewirkt jedoch auch, dass das Stickstoffatom keinen positiven mesomeren Effekt ausbilden kann. Die Reaktivität des Pyridinrings im Chinolinmolekül wird zu großen Teilen von dem negativen induktiven Effekt des Stickstoffatoms bestimmt. Sein Einfluss ist im Benzolring jedoch geringer.
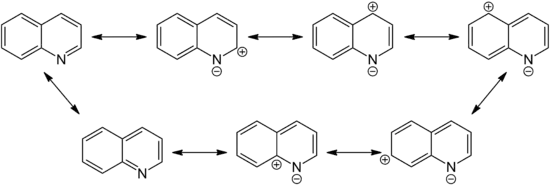
Chinolin ist über sieben mesomere Grenzstrukturen resonanzstabilisiert. Analog dem Naphthalin existieren zwei Grenzstrukturen, welche keinen zwitterionischen Charakter besitzen. Zusätzlich können jedoch fünf weitere zwitterionische Grenzstrukturen formuliert werden, welche dem Stickstoffatom eine negative Ladung zuweisen, wodurch die positive Ladung über das aromatische System verteilt wird. Die Lage der Ladung am Stickstoffatom steht im Einklang mit dessen höherer Elektronegativität im Vergleich zu Kohlenstoff.
Reaktionen
Elektrophile Substitutionen
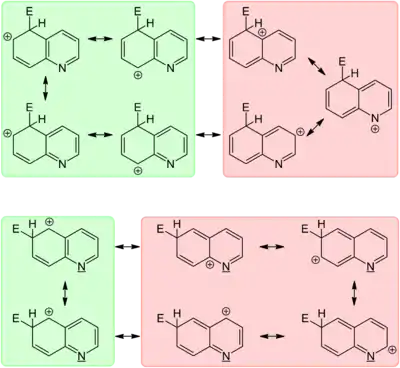
Im Vergleich zu Pyridin reagiert Chinolin leichter im Sinne einer elektrophilen aromatischen Substitution. Dieser Umstand ist der höheren mittleren Elektronendichte im aromatischen System geschuldet, die durch den relativ elektronenreicheren Benzolring der Verbindung hervorgerufen wird. Auf Grund der höheren Elektronendichte im Benzolring, finden elektrophile Substitutionen bevorzugt an diesem statt. Häufig sind im Reaktionsgemisch für elektrophile Substitutionen auch Brønsted- oder Lewis-Säuren anwesend. Diese können an das Stickstoffatom des Pyridinrings addieren und verursachen damit eine noch stärkere Desaktivierung des Pyridinrings.[5][29] Aus den beschriebenen elektronischen Gründen laufen elektrophile Substitutionen am schnellsten an den 5- und 8-Positionen des Chinolins ab. So wird als Nitrierungsprodukt eine Mischung aus gleichen Teilen von 5- und 8-Nitrochinolin erhalten, während weitere Isomere nur in untergeordnetem Maße gebildet werden.[52] Die Nitrierung von Chinolin weist hierbei eine schwächere Selektivität auf als jene des Isochinolins, bei der praktisch ausschließlich 5-Nitroisochinolin gebildet wird.[5]
Durch Sulfonierung mit Oleum bei moderater Temperatur wird das in 8-Position substituierte Chinolinderivat als Hauptprodukt und ferner das 5-Chinolylderivat erhalten. Da die elektrophile Substitution an diesen Positionen am schnellsten abläuft, handelt es sich hierbei um die kinetischen Reaktionsprodukte. Bei Erhitzung des Produktgemischs auf über 250 °C findet eine Isomerisierung zur thermodynamisch günstigeren Chinolin-6-sulfonsäure statt.[53]
Die Zusammensetzung des durch Halogenierung von Chinolin mit molekularen Halogenen erhaltene Produktspektrum unterliegt stark den Reaktionsbedingungen. Durch Bromierung in Schwefelsäure gehen jedoch meist die in 5- und 8-Position substituierten Chinolinderivate als Hauptprodukte aus der Reaktion hervor.[54] Durch Verwendung des Chinolinhydrobromids ist auch die Substitution am Pyridinring unter milden Reaktionsbedingungen möglich, welche in 3-Position abläuft.[55]
Nukleophile Substitutionen
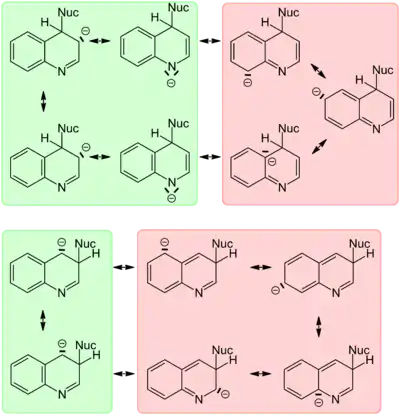
Viele aus der Pyridinchemie bekannte nukleophile Substitutionen laufen auch an Chinolin, bevorzugt an der elektronenarmen 2-Position des Pyridinrings, ab. Hierzu gehört Aminierung durch eine Tschitschibabin-Reaktion, bei welcher durch Verwendung von Kaliumamid als Nukleophil in flüssigem Ammoniak bei −66 °C das Amidion bevorzugt an die 2-Position von Chinolin addiert. Durch anschließende Oxidation mit Kaliumpermanganat kann 2-Aminochinolin freigesetzt werden.[56] Bei Erhöhung der Reaktionstemperatur auf −40 °C findet eine Isomerisierung zum thermodynamisch stabileren 4-substituierten Produkt statt.[57]
Chinolin kann durch Verwendung der zu Grunde liegenden Lithiumorganyle oftmals direkt alkyliert oder aryliert werden. Das nach wässriger Aufarbeitung der Reaktion als Zwischenprodukt entstehende Dihydrochinolinderivat kann thermisch rearomatisiert werden.[58] Sind gute Abgangsgruppen vorhanden, so ist analog dem Pyridin eine Reihe von ipso-Substitutionen an Chinolin bekannt. Substitutionen an der 3-Position weisen hierbei eher Charakteristika von ipso-Substitutionen an den entsprechenden Halogenaromaten auf, während solche an den 2- und 4-Positionen denen an Pyridin ähneln.[5]
Lithiierte Chinoline können aus den zu Grunde liegenden Halogenderivaten sowohl am Pyridin-[59][60] als auch am Benzolring[61] mittels kommerziell erhältlichen Lithiumorganylen wie n-Butyllithium hergestellt werden. Diese Reaktion steht in Konkurrenz zur oben beschriebenen Alkylierung, welche jedoch durch Reaktionsführung bei niedrigen Temperaturen in vielen Fällen weitgehend zurückgedrängt werden kann.[5] Die erhaltenen lithiierten Chinolinderivate können entweder direkt als Nukleophile verwendet oder zuvor auf ein anderes Metallion transmetalliert werden.
Oxidation und Reduktion
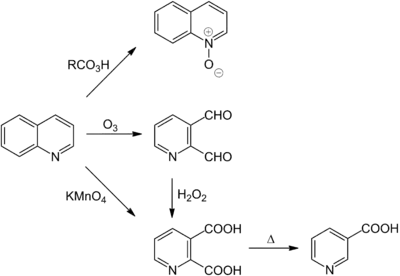
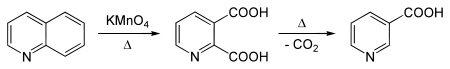
Analog dem Pyridin-N-oxid bildet auch Chinolin ein N-Oxid, welches durch Oxidation von Chinolin mit Peroxycarbonsäuren, oftmals Perbenzoesäure, hergestellt werden kann.[62] Unter stark oxidierenden Bedingungen tritt hingegen der oxidative Abbau des Benzol- oder des Pyridinrings ein. Welcher der Ringe dem Abbau unterworfen ist, hängt von den Reaktionsbedingungen ab. In der Regel ist hiervon der Benzolring betroffen,[5] wobei oftmals Chinolinsäure entsteht. Als Oxidationsmittel können Kaliumpermanganat,[63] Braunstein[64] oder rauchende Salpetersäure[64] dienen. Als effizienteste Methode gilt jedoch die elektrochemische Oxidation.[5][64] Die Ozonolyse von Chinolin liefert Pyridin-2,3-dialdehyd[65], welcher durch anschließende Oxidation mit Wasserstoffperoxid wiederum zu Chinolinsäure oxidiert werden kann.[66]
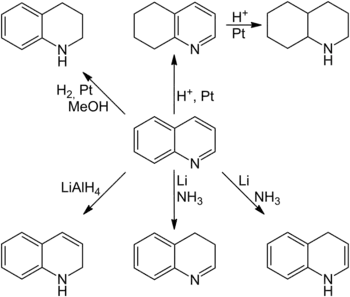
Je nach Reaktionsbedingung kann sowohl der Benzol- als auch der Pyridinring selektiv hydriert werden. Zur Hydrierung des Pyridinrings wird klassisch molekularer Wasserstoff in Methanol bei Raumtemperatur am Platinkatalysator verwendet.[5] Es sind jedoch auch Hydrierungen durch Natriumcyanoborhydrid[67] sowie Natriumborhydrid[68] und Zinkborhydrid[69] in Gegenwart von Nickel(II)-chlorid bekannt. Die selektive Hydrierung des Benzolrings gelingt durch Umsetzung mit Wasserstoff in starken Säuren am Platinkatalysator.[70] Durch längere Reaktionsdauer kann unter diesen Bedingungen auch das vollständig gesättigte Decahydrochinolin erhalten werden.[5] Auch dihydrierte Chinolinderivate sind synthetisch aus Chinolin zugänglich. 1,2-Dihydrochinolin kann durch Reduktion mit Lithiumaluminiumhydrid[71] hergestellt werden, während 1,4-[72] und 3,4-Dihydrochinolin[5] sind durch Reduktion mit elementarem Lithium in flüssigem Ammoniak zugänglich sind. Dihydrierte Chinolinderivate neigen jedoch häufig zu Isomerisierung der Doppelbindung und können deshalb manchmal nicht isoliert werden, sondern treten nur als Zwischenprodukte einer Reaktion auf.
Verwendung
Ullmanns Enzyklopädie der Technischen Chemie beziffert im Jahre 2005 die Weltjahresproduktion von Chinolin mit 2000 Tonnen.[11] In Analogie zum steigenden Bedarf ähnlicher heterocyclischer Synthesebausteine ist jedoch davon auszugehen, dass die Produktionskapazität zwischenzeitlich erhöht wurde. In der chemischen Industrie besitzt Chinolin weite Verwendungsbereiche. Es ist sowohl ein bedeutender Grundstoff zur Herstellung von Arzneimitteln als auch von Herbiziden und Fungiziden. Des Weiteren wird es auch als basischer Katalysator beispielsweise in der pharmazeutischen Industrie verwendet.[73][74]
Zu den Hauptverwendungen von Chinolin gehört die Herstellung von 8-Hydroxychinolin, einem Komplexbildner, der als Desinfektionsmittel und Antimykotikum verwendet wird. Die Synthese von 8-Hydroxychinolin gelingt durch Sulfonierung von Chinolin zu Chinolin-8-sulfonsäure und anschließender ipso-Hydroxylierung mit heißer Natronlauge.[75] Des Weiteren dient Chinolin der Herstellung von Chinolinsäure, welche ein bedeutender Grundstoff zur Herstellung von Herbiziden wie Imazapyr[76] ist. Chinolin ist außerdem ein Grundstoff zur industriellen Synthese von Nicotinsäure (Vitamin B3),[77] welche durch Decarboxylierung von Chinolinsäure zugänglich ist. Als klassischer Abbau dient hierbei die Oxidation mittels starker Oxidationsmittel wie Kaliumpermanganat,[78] moderne industrielle Verfahren verwenden jedoch günstigere Oxidationsmittel.[79][80]
Auch teilweise hydrierte Chinolinderivate dienen der Herstellung pharmazeutischer Wirkstoffe und Antibiotika.[81][82] 2-Hydroxychinolin, welches oxidativ durch Hypochlorige Säure oder enzymatische Hydroxylierung aus Chinolin zugänglich ist, stellt ebenfalls einen Grundstoff zur Herstellung von beispielsweise herzwirksamen Arzneistoffen oder Antihistaminika dar.[11] Außerdem ist es Grundstoff zur industriellen Synthese von Cyaninfarbstoffen.
Im chemischen Labor oder chemischen Anlagen kann Chinolin als gutes Lösungsmittel und sehr gutes Extraktionsmittel für polycyclische aromatische Kohlenwasserstoffe eingesetzt werden. Manchmal werden hierzu auch Gemische mit Isochinolin verwendet. Chinolin findet des Weiteren als Korrosionsinhibitor und als säurebindende Base in chemischen Reaktionen Einsatz.[11] In der Palladium-katalysierten Hydrierung von Alkinen dient Chinolin zur partiellen Desaktivierung (Vergiftung) des Katalysators (sogenannter Lindlar-Katalysator).[83][84] Ein solchermaßen desaktivierter Katalysator ermöglicht die einfache Hydrierung des Alkins zum zu Grunde liegenden Alkens und verhindert die zweifache Hydrierung zum Alkan.[85] Auch zur Rosenmund-Reduktion, einer ebenfalls Palladium-katalysierten Reduktion von Carbonsäurechloriden zu Aldehyden, werden mit Chinolin desaktivierte Katalysatoren verwendet.[86]
Gefahrenhinweise
Chinolin ist als toxisch und umweltgefährdend eingestuft und darf nur bei starker Entlüftung und nur mit geeigneten Schutzhandschuhen gehandhabt werden. Die Freisetzung des Stoffes in die Umwelt ist zu verhindern.[4] Es bestehen hinreichende Anhaltspunkte, dass die Exposition eines Menschen mit Chinolin Krebs erzeugen kann. Des Weiteren besteht der begründete Verdacht auf eine erbgutverändernde Wirkung.[4] Chinolin ist als wassergefährdend Klasse 2 eingestuft.[4]
Mit Luft bildet Chinolin ab seinem Flammpunkt von 101 °C zündfähige Luft-Dampf-Gemische. Der Explosionsbereich liegt zwischen 1 Vol.‑% (54 g/m³) als untere Explosionsgrenze (UEG) und 7 Vol.‑% (376 g/m³) als obere Explosionsgrenze (OEG).[4] Die Zündtemperatur beträgt 480 °C.[87] Der Stoff fällt somit in die Temperaturklasse T1. Bei Bränden können nitrose Gase als Zersetzungsprodukte auftreten.[4] Die elektrische Leitfähigkeit ist mit 2,2·10−6 S·m−1 eher gering.[88]
Toxikologie
Chinolin weist bei peroraler Aufnahme durch Ratten eine mittlere letale Dosis von 270 mg·kg−1 auf,[9] bei dermaler Exposition liegt sie hingegen bei 1400 mg·kg−1.[11] In einer Untersuchung an Ratten trat nach siebenstündiger Inhalation von Chinolin-gesättigter Luft der Tod fast aller Versuchstiere ein. Als akute Symptome der Exposition traten Ataxie und Gänsehaut auf. Bei den toten Versuchstieren wurden Blutungen und Ödeme im Verdauungstrakt festgestellt, welche auf die Chinolinexposition zurückzuführen sind.[11] Bei dermaler Exposition bildeten sich in Untersuchungen mit Kaninchen leichte bis moderate Ödeme und Rötungen, die Haut regenerierte sich jedoch nach Beendigung der Versuchsreihe wieder vollständig. Es wurde auch eine leichte bis moderate, jedoch ebenfalls reversible, augenreizende Wirkung beobachtet.[11]
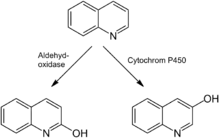
Bei peroraler Aufnahme ergab sich im Langzeitversuch bei Ratten und Mäusen eine karzinogene Wirkung, welche sich in der Ausbildung von Leberzellkarzinomen und Angiosarkomen manifestierte.[89][90] Im Gegensatz hierzu traten diese Befunde bei Meerschweinchen und Hamstern nicht auf.[90] Eine geringe karzinogene Wirkung bei dermaler Exposition zeigte sich an Mäusen. Nach einer Untersuchung ist die zur Ausbildung von Karzinomen nötige Menge an Chinolin 250–300 mal höher als bei dem starken Karzinogen Benzapyren.[91] Für die karzinogene Wirkung ist jedoch nicht Chinolin selbst, sondern dessen Metabolite verantwortlich. Es sind verschiedene Abbaupfade von Chinolin und dessen Derivaten unter aeroben und anaeroben Bedingungen bekannt. Welcher Pfad beschritten wird, ist vom betrachteten Organismus abhängig.[92] Der erste Abbauschritt besteht meist aus einer Oxidation des Aromaten, welche beispielsweise durch Aldehydoxidase zu 2-Hydroxychinolin oder durch Cytochrom-P450-Proteine zu 3-Hydroxychinolin führt.[93] Säugetiere scheiden die oxidierten Abbauprodukte in kurzer Zeit über den Magen-Darm-Trakt aus.[94][95]
Chinolin in der Umwelt
Chinolin wurde in Spuren in der Umgebung Aluminium-verhüttender Betriebe mit angeschlossenen Kokereien sowie Kohle-verarbeitender Betriebe gefunden, was auf das Vorkommen von Chinolinderivaten in der Kohle zurückzuführen ist. Auch bei der Verarbeitung von Steinkohlenteer und Teeröl werden Spuren von Chinolin freigesetzt. Teeröl wird oder wurde als fäulnishemmendes Mittel zur Imprägnierung von Holz, beispielsweise für Bahnschwellen oder Telegrafenmasten verwendet, wodurch Chinolin auch entfernt von entsprechender Industrie in die Umgebung freigesetzt werden kann. Des Weiteren kann Chinolin als Spurengas bei der unvollständigen Verbrennung organischer stickstoffhaltiger Verbindungen entstehen.[21][96]
Aus kontaminiertem Erdreich wird Chinolin durch Wasser in kurzer Zeit ausgewaschen und durch Bakterien und Huminsäuren abgebaut.[21] In der Regel herrschen hierzu in oberflächennahem Wasser günstigere Bedingungen, während der Abbau in Tiefenwasser auf Grund fehlender geeigneter Organismen und ungünstiger chemischer Bedingungen (unter anderem auf Grund der geringen Sauerstoffkonzentration) nur langsam vonstattengeht. Oberflächennah oder atmosphärisch vorhandenes Chinolin unterliegt außerdem der Zersetzung durch Photolyse. Dieser Abbaupfad ist stark abhängig von der Photonendichte, dem pH-Wert und der Temperatur. Je nach Bedingungen liegt die Halbwertszeit von Chinolin durch photolytische Degradation zwischen 21 und 160 Tagen.[21] Die Verbindung besitzt nur ein geringes Potential zur Bioakkumulation, da Chinolin von Bakterien, Fischen und Säugetieren rasch abgebaut wird. Modellrechnungen zur Verbreitung von Chinolin ergaben, dass die Verbindung im Wesentlichen durch Wasser transportiert wird. Der Transport durch die Atmosphäre tritt auf Grund des geringen Dampfdrucks in den Hintergrund.[96]
Nachweis
Das UV/Vis-Spektrum von Chinolin weist drei Absorptionsbanden auf. Diese resultieren aus π→π*- und n→π*-Übergängen und treten bei Wellenlängen von 226 nm (Extinktionskoeffizient ε = 35.500 l·(mol·cm)−1), 270 nm (ε = 3880 l·(mol·cm)−1) und 313 nm (ε = 2360 l·(mol·cm)−1) auf.[97] Das Infrarotspektrum von Chinolin weist eine Vielzahl von Absorptionsbanden auf. Charakteristische starke und sehr starke Banden treten bei 3333, 1034, 941, 808, 787, 760 und 740 cm−1 auf. Des Weiteren existieren acht weitere eng beieinander liegende starke Absorptionsbanden zwischen 1629 und 1319 cm−1.[98]
Die Protonensignale im lösungsmittelfreien 1H-NMR-Spektrum von Chinolin liegen ausnahmslos in einem Bereich, der für aromatische Protonen charakteristisch ist. Innerhalb dieses Bereiches weisen sie jedoch in Relation zu Benzol teils ausgeprägte Verschiebung zu tieferen Feldern auf und sind ein Ausdruck der verminderten Elektronendichte an diesen Wasserstoffatomen. Das Spektrum zeigt sieben Signale korrespondierend mit den sieben chemisch verschiedenen Protonen im Molekül. Da jedes Signal ein Proton repräsentiert, weisen die Signale gleiche Flächenintegrale auf. Das Signal bei tiefstem Feld resultiert von dem Proton in 2-Position δ(2-H) = 8,8 ppm, gefolgt von den Protonen in 8- (δ(8-H) = 8,1 ppm) und 4-Position (δ(4-H) = 8,0 ppm). Die weiteren Protonensignale liegen im Bereich 7,7 und 7,3 ppm. Die größeren chemischen Verschiebungen der Protonen im Vergleich zum Kohlenstoff-Analogon Naphthalin resultieren aus der geringeren Elektronendichte im aromatischen System und korrespondieren relativ mit den niedrigeren Elektronendichten in diesen Positionen, welche aus den mesomeren Grenzstrukturen abgeleitet werden können. Entsprechend der Anzahl der Kohlenstoffatome treten im 13C-NMR-Spektrum neun Signale im Bereich zwischen 122 und 151 ppm auf. Die chemischen Verschiebungen der 13C-Kerne verhalten sich hierbei analog den Protonensignalen. Die beiden Positionen mit niedriger Elektronendichte, die sich in Nachbarschaft des Stickstoffatoms finden, weisen die höchsten Tieffeldverschiebungen auf (151 beziehungsweise 149 ppm).[97]
Literatur
- T. Eicher, S. Hauptmann: The Chemistry of Heterocycles. 2. Auflage. Wiley-VCH, Weinheim 2003, ISBN 3-527-30720-6.
- J. A. Joule, K. Mills: Heterocyclic Chemistry. 3. Auflage. Blackwell Science, Oxford 2004, ISBN 0-632-05453-0.
- D. T. Davies: Basistexte Chemie: Aromatische Heterocyclen. 1. Auflage. Wiley-VCH, Weinheim 1995, ISBN 3-527-29289-6.
Weblinks
Einzelnachweise
- Eintrag zu Chinolin. In: Römpp Online. Georg Thieme Verlag, abgerufen am 10. November 2014.
- O. Eckstein: Ueber Chinolin-chlorhydrat und die Einwirkung von Säurechloriden auf Chinolin. In: Chem. Ber., 1906, 39, S. 2135–2138; doi:10.1002/cber.190603902173.
- David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press/Taylor and Francis, Boca Raton, FL, Physical Constants of Organic Compounds, S. 3-454.
- Eintrag zu Chinolin in der GESTIS-Stoffdatenbank des IFA, abgerufen am 8. Januar 2021. (JavaScript erforderlich)
- J. A. Joules, K. Mills: Heterocyclic Chemistry. 5. Auflage. Blackwell Publishing, Chichester 2010, ISBN 978-1-4051-9365-8, S. 177–199.
- A. D. Buckingham, J. Y. H. Chau, H. C. Freeman, R. J. W. Le Fèvre, D. A. A. S. Narayana Rao, J. Tardif: The dipole moments of pyridine, quinoline, and isoquinoline as vapours and as solutes. In: J. Chem. Soc., 1956, S. 1405–1411; doi:10.1039/JR9560001405.
- W. H. F. Sasse: Synthetical applications of activated metal catalysts. Part VIII. The action of degassed Raney nickel on quinoline and some of its derivatives. In: J. Chem. Soc., 1960, S. 526–533; doi:10.1039/JR9600000526.
- Eintrag zu Quinoline im Classification and Labelling Inventory der Europäischen Chemikalienagentur (ECHA), abgerufen am 1. Februar 2016. Hersteller bzw. Inverkehrbringer können die harmonisierte Einstufung und Kennzeichnung erweitern.
- Datenblatt Chinolin (PDF) bei Merck, abgerufen am 5. September 2010.
- W. V. Steele, D. G. Archer, R. D. Chirico, W. B. Collier, I. A. Hossenlopp, A. Nguyen, N. K. Smith, B. E. Gammon: The thermodynamic properties of quinoline and isoquinoline. In: J. Chem. Thermodynamics, 1988, S. 1233–1264; doi:10.1016/0021-9614(88)90161-9.
- G. Collin, H. Höke: Quinoline and Isoquinoline. In: Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim 2005.
- Ch. Gerhardt: Untersuchungen über die organischen Basen. In: Annalen der Chemie und Pharmacie, 1942, 42, S. 310–313; doi:10.1002/jlac.18420420310.
- Ch. Gerhardt: Untersuchungen über die chemische Classification der organischen Substanzen. In: J. Prakt. Chem., 1843, 28, S. 65–100; doi:10.1002/prac.18430280112.
- Monatsbericht: Chinolin. In: Archiv der Pharmazie, 1882, 20, S. 50–73; doi:10.1002/ardp.18822200109.
- Paul Rabe: Untersuchungen in der Pyridin- und Chinolinreihe. In: J. Prakt. Chem., 1938, 151, S. 65–81; doi:10.1002/prac.19381510203.
- W. Königs: Oxydationsprodukte des Cinchonins. In: Chem. Ber., 1879, 12, S. 79–101; doi:10.1002/cber.18790120130.
- S. Hoogewerff, W. A. van Dorp: Sur la quinoléine du goudron de houille et des alcaloïdes du quinquina, et sur leur oxydation au moyen du permanganate de potassium. In: Rec. Trav. Chim., 1882, 1, S. 1–17; doi:10.1002/recl.18820010501.
- B. Aronheim: Synthese des Naphthalin. In: Chem. Ber., 1873, 6, S. 67–68; doi:10.1002/cber.18730060125.
- W. Koenigs: Synthese des Chinolins aus Allylanilin. In: Chem. Ber., 1879, 12, S. 453; doi:10.1002/cber.187901201128.
- M. Hesse: Alkaloide. 1. Auflage. Verlag Helvetica Chimica Acta, Zürich 2000, ISBN 3-906390-19-5, S. 375–378.
- Toxological Review of Quinoline (PDF; 175 kB). (PDF) Environmental Protection Agency, September 2001.
- D. Hellwinkel: Die systematische Nomenklatur der Organischen Chemie. 4. Auflage. Springer Verlag, Berlin 1998, ISBN 3-540-63221-2.
- A. Gossauer: Struktur und Reaktivität der Biomoleküle, 2006, S. 488, Wiley-VCH Weinheim, ISBN 3-906390-29-2.
- G. Lunge: Die Destillation des Steinkohlentheers und die Verarbeitung der damit zusammenhängenden Nebenproducte, Friedrich Vieweg und Sohn, Braunschweig, 1867.
- S. Shimizu, N. Watanabe, T. Kataoka, T. Shoji, N. Abe, S. Morishita, H. Ichimura: Pyridine and Pyridine Derivatives. In: Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim 2005.
- Europäisches Patent: EP 100109, 1983, Rütgerswerke.
- Japanisches Patent: JP 86161265, 1986, Sumikin Coke & Chemicals.
- Z. H. Skraup: Eine Synthese des Chinolins. In: Chem. Ber., 1880, 13, S. 2086–2087; doi:10.1002/cber.188001302195.
- D. T. Davies: Basistexte Chemie: Aromatische Heterocyclen. 1. Auflage. Wiley-VCH, Weinheim 1995, ISBN 3-527-29289-6, S. 46–52.
- O. Doebner, W. v. Miller: Ueber eine dem Chinolin homologe Base. In: Chem. Ber., 1881, 14, S. 2812–2817; doi:10.1002/cber.188101402258.
- F. W. Bergstrom, Heterocyclic Nitrogen Compounds. Part IIA. Hexacyclic Compounds: Pyridine, Quinoline, and Isoquinoline. In: Chem. Rev., 1944, 35, S. 77–277; doi:10.1021/cr60111a001.
- A. Shaabani, E. Soleimani, Z. Badri: Triflouroacetic Acid as an Efficient Catalyst for the Synthesis of Quinoline. In: Synth. Commun., 2007, 37, S. 629–635; doi:10.1080/00397910601055230.
- C.-S. Jia, Z. Zhang, S.-J. Tu, G.-W. Wang: Rapid and efficient synthesis of poly-substituted quinolines assisted by p-toluene sulphonic acid under solvent-free conditions: comparative study of microwave irradiation versus conventional heating. In: Org. Biomol. Chem., 2006, 4, S. 104–110; doi:10.1039/b513721g.
- J. Wu, H.-G. Xia, K. Gao: Molecular iodine: a highly efficient catalyst in the synthesis of quinolines via Friedländer annulation. In: Org. Biomol. Chem., 2006, 4, S. 126–129; doi:10.1039/b514635f.
- R. Varala, R. Enugala, S. R. Adapa: Efficient and Rapid Friedlander Synthesis of Functionalized Quinolines Catalyzed by Neodymium(III) Nitrate Hexahydrate. In: Synthesis, 2006, S. 3825–3830; doi:10.1055/s-2006-950296.
- R. H. Manske: The Chemistry of Quinolines. In: Chem. Rev., 1942, 30, S. 113–144; doi:10.1021/cr60095a006.
- V. V. Kouznetsov, L. Y. V. Méndez, L. Y. V. Gómez: Recent Progress in the Synthesis of Quinolines. In: Curr. Org. Chem., 2005, 9, S. 141–161; doi:10.1002/chin.200516245.
- R. H. Reitsema: The Chemistry of 4-Hydroxyquinolines. In: Chem. Rev., 1948, 43, S. 43–68; doi:10.1021/cr60134a002.
- R. Camps: Synthese von alpha- und gamma-Oxychinolinen. In: Chem. Ber., 1899, 22, S. 3228–3234; doi:10.1002/cber.18990320389.
- L. Knorr: Synthetische Versuche mit dem Acetessigester. In: Liebigs Ann., 1886, 236, S. 69–115; doi:10.1002/jlac.18862360105.
- R. G. Gould, W. A. Jacobs: The Synthesis of Certain Substituted Quinolines and 5,6-Benzoquinolines. In: J. Am. Chem. Soc., 1939, 61, S. 2890–2895; doi:10.1021/ja01265a088.
- P. M. Dewick: Medicinal Natural Products: A Biosynthetic Approach. 1. Auflage. John Wiley & Sons, New York 2009, ISBN 0-470-74167-8, S. 380–397.
- S. Malanowski: Vapour Pressures and Boiling Temperatures of Some Quinoline Bases. In: Bull. Acad. Pol. Sci. Ser. Sci. Chim., 1961, 9, S. 71–76.
- David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press/Taylor and Francis, Boca Raton, FL, Fluid Properties, S. 6-675.
- F. Glaser, H. Ruland: Untersuchungen über Dampfdruckkurven und kritische Daten einiger technisch wichtiger organischer Substanzen. In: Chem. Ing. Techn., 1957, 29, S. 772.
- David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press/Taylor and Francis, Boca Raton, FL, Thermochemistry, Electrochemistry, and Solution Chemistry, S. 5-38.
- David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press/Taylor and Francis, Boca Raton, FL, Fluid Properties, S. 6-212.
- David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press/Taylor and Francis, Boca Raton, FL, Fluid Properties, S. 6-223.
- J. E. Davies, A. D. Bond: Quinoline. In: Acta Cryst., 2001, E57, S. o947–o949; doi:10.1107/S1600536801014891.
- David R. Lide (Hrsg.): CRC Handbook of Chemistry and Physics. 90. Auflage. (Internet-Version: 2010), CRC Press/Taylor and Francis, Boca Raton, FL, Physical Constants of Organic Compounds, S. 3-448.
- J. Buddrus: Grundlagen der Organischen Chemie. S. 337. 3. Auflage., de Gruyter Verlag, Berlin 2003, ISBN 3-11-014683-5.
- M. W. Austin, J. H. Ridd: The kinetics and mechanism of heteroaromatic nitration. Part I. Quinoline. In: J. Chem. Soc., 1963, S. 4204–4210; doi:10.1039/JR9630004204.
- G. E. McCasland: The Preparation of 8-Qinolinesulfonic acid. In: J. Org. Chem., 1946, 11, S. 277–280; doi:10.1021/jo01173a010.
- J. L. Butler, M. Gordon: A reinvestigation of known bromination reactions of quinoline. In: J. Heterocycl. Chem., 1975, 12, S. 1015–1020; doi:10.1002/jhet.5570120539.
- T. J. Kress, S. M. Constantino: Selective brominations in nitrobenzene. A convenient synthesis of 3-brornoquinoline, 4-bromoisoquinoline, and 4-phenyl-5-bromopyrimidine. In: J. Heterocycl. Chem., 1973, 10, S. 409–410; doi:10.1002/jhet.5570100326.
- H. van der Plas: Oxidative Amino-Dehydrogenation of Azines. In: Adv. Heterocycl. Chem., 2004, 86, S. 1–40; doi:10.1016/S0065-2725(03)86001-4.
- J. A. Zoltewicz, L. S. Helmick, T. M. Oestreich, R. W. King, P. E. Kandetzki: Addition of amide ion to isoquinoline and quinoline in liquid ammonia. Nuclear magnetic resonance spectra of anionic σ-complexes. In: J. Org. Chem., 1973, 38, S. 1947–1949; doi:10.1021/jo00950a036.
- T. J. Geissmann, M. J. Schlatter, I. D. Webb, J. D. Roberts: The Synthesis of some Intermediates for the Use in the Preparation of Analogs of Salicylaldehyde ethylenediimine cobalt ("Salcomine"). In: J. Org. Chem., 1946, 11, S. 741–750; doi:10.1021/jo01176a015.
- M. Ishikura, T. Mano, I. Oda, M. Terashima: An Alternative Synthesis of Dialkylpyridylboranes. In: Heterocycles, 1984, S. 4271–4274; doi:10.3987/R-1984-11-2471.
- H. Gilman, T. Soddy: Notes - Some Organolithium Compounds of Quinoline and 2-Phenylquinoline. In: J. Org. Chem., 1958, 23, S. 1584–1585; doi:10.1021/jo01104a627.
- J. B. Wommack, T. G. Barbee Jr., D. J. Thoennes, M. A. McDonald, D. E. Pearson: The synthesis of quinoline- and isoquinolinecarboxaldehydes. In: J. Heterocycl. Chem, 1969, 6, S. 243–245; doi:10.1002/jhet.5570060217.
- J. Meisenheimer: Über Pyridin-, Chinolin- und Isochinolin-N-oxyd. In: Ber. Dtsch. Chem. Ges., 1926, 59, S. 1848–1853; doi:10.1002/cber.19260590828.
- C. F. Koelsch, A. F. Steinhauer: Synthesis, Nitration, and Oxidation of 3-Azafluoranthene. In: J. Org. Chem., 1953, 18, S. 1516–1522; doi:10.1021/jo50017a010.
- J. C. Cochran, W. F. Little: Electrolytic Oxidation of Some Substituted Quinolines to Quinolinic Acids and Acylations with Substituted Quinolinic Anhydrides. In: J. Org. Chem., 1961, 21, S. 808–811; doi:10.1021/jo01062a039.
- G. Queguiner, P. Pastour: Synthèse dans la série de la pyridine - I - Les diformylpyridines. In: Bull. Soc. Chim. Fr., 1968, 10, S. 4117–4121; OCLC 491832299.
- C. O’Murchu: Ozonolysis of Quinolines: A Versatile Synthesis of Polyfunctional Pyridines. In: Synthesis, 1989, 11, S. 880–882; doi:10.1055/s-1989-27423.
- G. R. Girard, W. E. Bondinell, L. M. Hillegass, K. G. Holden, R. G. Pendleton, I. Uzinskas: Tetrahydro thiadiazolo isoquinolines: synthesis and inhibition of phenylethanolamine-N-methyltransferase. In: J. Med. Chem., 1989, 32, S. 1566–1571; doi:10.1021/jm00127a027.
- A. Nose, T. Kudo: Reduction of Heterocyclic Compounds. II. Reduction of Heterocyclic Compounds with Sodium Borohydride-Transition Metal Salt Systems. In: Chem. Pharm. Bull., 1984, 32, S. 2421–2425; Abstract.
- B. C. Ranua, U. Jana, A. Sarkara: Regioselective Reduction of Quinolines and Related Systems to 1,2,3,4-Tetrahydro Derivatives with Zinc Borohydride. In: Synth. Commun., 1998, 28, S. 485–492; doi:10.1080/00397919808005103.
- G. L. Patrick: Synthesis of (±)-[4aα,4bβ,10bβ,12aα]-9-halogeno-2-methyl-1,2,3,4,4a,4b,5,6,10b,11,12,12a-dodecahydronaphtho[2,1-f]isoquinolines. In: J. Chem. Soc., Perkin Trans. 1, 1995, S. 1273–1279; doi:10.1039/P19950001273.
- E. A. Braude, J. Hannah, Sir R. Linstead: Hydrogen transfer. Part XVI. Dihydrides of nitrogenous heterocycles as hydrogen donors. In: J. Chem. Soc., 1960, S. 3249–3257; doi:10.1039/JR9600003249.
- Arthur Birch, P. G. Lehmann: 1,4-Dihydrochinolin. In: Tetrahedron Lett., 1974, 15, S. 2395–2396; doi:10.1016/S0040-4039(01)92265-8.
- M. F. Depompei, A. Hlynsky, Diamond Shamrock, US 4 281 125, 1980.
- J. P. Senet, G. Sennvey, G. Wooden, Société Nationale des Poudres et Explosifs, EP 249 556, 1987.
- N. N. Woroshtzow, J. M. Kogan: Über die Wirkung von schwefliger Säure und ihren Salzen auf Chinolin-Derivate. In: Ber. Dtsch. Chem. Ges., 1930, S. 2354–2362; doi:10.1002/cber.19300630878.
- P. L. Orwick, A. R. Templeton, American Cyanamid, EP 41 623, 41 624, 1981.
- A. Streitwieser, C. H. Heathcock, E. M. Kosower: Organische Chemie. 2. Auflage. Wiley-VCH, Weinheim 1994, ISBN 3-527-29005-2, S. 1227.
- S. Hoogewerff, W. A. van Dorp: Ueber die Oxydation von Chinolin vermittelst Kaliumpermanganat. In: Chem. Ber., 1879, 12, S. 747–748; doi:10.1002/cber.187901201208.
- W. Orth, E. Pastorek, W. Fickert, Rütgerswerke, EP 149 857, 1984.
- R. W. J. Rebhahn, J. E. Kassner, R. E. Werner, Hilton-Davis Chemical, US 4 537 971, 1985.
- S. Murahashi, Sumitomo Chemical, JP 87 212 363, 1986.
- R. Lattrell, W. Dürckheimer, R. Kirstetter, Hoechst, DE 3 706020, 1987.
- H. Lindlar, R. Dubuis: Palladium Catalyst for Partial Reduction of Acetylenes In: Organic Syntheses. 46, 1966, S. 89, doi:10.15227/orgsyn.046.0089; Coll. Vol. 5, 1973, S. 880 (PDF).
- H. Lindlar: Ein neuer Katalysator für selektive Hydrierungen. In: Helv. Chim. Acta, 1952, 35, S. 446–450; doi:10.1002/hlca.19520350205.
- J. Buddrus: Grundlagen der Organischen Chemie. 3. Auflage. de Gruyter Verlag, Berlin 2003, ISBN 3-11-014683-5, S. 295.
- J. Buddrus: Grundlagen der Organischen Chemie. 3. Auflage. de Gruyter Verlag, Berlin 2003, ISBN 3-11-014683-5, S. 538.
- E. Brandes, W. Möller: Sicherheitstechnische Kenngrößen – Band 1: Brennbare Flüssigkeiten und Gase, Wirtschaftsverlag NW – Verlag für neue Wissenschaft, Bremerhaven 2003.
- Technische Regel für Betriebssicherheit – TRBS 2153, BG RCI Merkblatt T033 Vermeidung von Zündgefahren infolge elektrostatischer Aufladungen, Stand April 2009. Jedermann-Verlag, Heidelberg.
- K. Hirao, Y. Shinohara, H. Tsuda, S. Fukushima, M. Takahashi, N. Ito: Carcinogenic Activity of Quinoline on Rat Liver. In: Cancer Res., 1976, 36, S. 329–335; PMID 177193 (Volltextzugriff (PDF); PDF; 1,3 MB).
- Y. Shinohara, T. Ogiso, M. Hananouchi, K. Nakanishi, T. Yoshimura, N. Ito: Effect of various Factors on the Induction of Liver Tumors in Animals by Quinoline. In: GANN, 1977, 68, S. 785–796; PMID 598648.
- E. J. LaVoie, A. Shigematsu, E. A. Adams, J. Rigotty, D. Hoffmann: Tumor-initiating activity of quinoline and methylated quinolines on the skin of SENCAR mice. In: Cancer Lett., 1984, 22, S. 269–273; doi:10.1016/0304-3835(84)90162-9; PMID 6324986.
- M. M. Ramirez-Corredores, A. P. Borole: Studies in Surface Science and Catalysis 164: Biocatalysis in Oil Refining. 1. Auflage. Elsevier Verlag, New York 2007, ISBN 0-444-52212-3, S. 155–160.
- P. Jenner, B. Testa: Concepts in Drug Metabolism: Part A. 3. Auflage. Marcel Dekker, New York 1980, ISBN 0-8247-6906-6, S. 115.
- L. Novack, B. B. Brodie: Quinoline and its transformation products found in urine. In: J. Biol. Chem., 1950, 187, S. 787–792 (Volltextzugriff).
- J. T. Smith, R. N. Williams: Studies in detoxication. 65. The metabolism of quinoline. New metabolites of quinoline, with observations on the metabolism of 3-, 5- and 6-hydroxyquinoline and 2:4-dihydroxyquinoline. In: Biochem. J., 1955, 60, S. 284–290; PMC 1215695 (freier Volltext).
- Untersuchungsbericht des Staates Kanada
- J. A. Joule, K. Mills: Heterocyclic Chemistry. 3. Auflage. Blackwell Science, Oxford 2004, ISBN 0-632-05453-0, S. 14–16.
- H. Tschammler, H. Krischai: Chinolin-m-Kresol, ein stark negatives System. In: Monatsh. Chem., 1951, 82, S. 259–270; doi:10.1007/BF00899511.