Uran
Uran (benannt nach dem Planeten Uranus) ist ein chemisches Element mit dem Elementsymbol U und der Ordnungszahl 92. Im Periodensystem steht es in der Gruppe der Actinoide (7. Periode, f-Block). Uran ist ein Metall, dessen sämtliche Isotope radioaktiv sind. Natürlich in Mineralen auftretendes Uran besteht zu etwa 99,3 % aus dem Isotop 238U und zu 0,7 % aus 235U.
| Eigenschaften | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allgemein | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Name, Symbol, Ordnungszahl | Uran, U, 92 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Elementkategorie | Actinoide | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Gruppe, Periode, Block | Ac, 7, f | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Aussehen | Silberweiß | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CAS-Nummer | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| EG-Nummer | 231-170-6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ECHA-InfoCard | 100.028.336 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Massenanteil an der Erdhülle | 3,2 ppm[1] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomar [2] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atommasse | 238,02891(3)[3] u | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomradius | 138,5 (α-Uran)[1] pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Kovalenter Radius | 142 pm | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Elektronenkonfiguration | [Rn] 5f3 6d1 7s2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1. Ionisierungsenergie | 6.19405(6) eV[4] ≈ 597.63 kJ/mol[5] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2. Ionisierungsenergie | 11.6(4) eV[4] ≈ 1120 kJ/mol[5] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3. Ionisierungsenergie | 19.8(3) eV[4] ≈ 1910 kJ/mol[5] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4. Ionisierungsenergie | 36.7(1,0) eV[4] ≈ 3540 kJ/mol[5] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5. Ionisierungsenergie | 46.0(1,9) eV[4] ≈ 4440 kJ/mol[5] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Physikalisch [6] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Aggregatzustand | fest | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Modifikationen | 3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Kristallstruktur | orthorhombisch (Raumgruppe Cmcm (Raumgruppen-Nr. 63)) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Dichte | 19,16 g/cm3[7] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mohshärte | 2,5–3[1] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Magnetismus | paramagnetisch (χm = 4,1 · 10−4)[8] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Schmelzpunkt | 1406 K (1133[7] °C) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Siedepunkt | 4203 K[7] (3930 °C) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molares Volumen | 12,49 · 10−6 m3·mol−1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Verdampfungsenthalpie | 417,1 kJ/mol[7] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Schmelzenthalpie | 15,5[7] kJ·mol−1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Schallgeschwindigkeit | ~3400 (long.), ~2000 (trans.) m·s−1 bei 293 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Spezifische Wärmekapazität | 116[1] J·kg−1·K−1 bei 298 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Elektrische Leitfähigkeit | 3,24 · 106[1] A·V−1·m−1 bei 293 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Wärmeleitfähigkeit | 27,6[1] W·m−1·K−1 bei 300 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Chemisch [9] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oxidationszustände | +3, +4, +5, +6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Normalpotential | −1,660 V (U3+ + 3 e− → U) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Elektronegativität | 1,38 (Pauling-Skala) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Isotope | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Weitere Isotope siehe Liste der Isotope | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Gefahren- und Sicherheitshinweise | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

Radioaktiv | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Soweit möglich und gebräuchlich, werden SI-Einheiten verwendet. Wenn nicht anders vermerkt, gelten die angegebenen Daten bei Standardbedingungen. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||


Eine besondere Bedeutung erhielt Uran nach der Entdeckung der Kernspaltung im Jahre 1938. Das Uranisotop 235U ist durch thermische Neutronen spaltbar und damit – neben dem äußerst seltenen, aber aus Uran erzeugbaren Plutonium-Isotop 239Pu – das einzige natürlich vorkommende Nuklid, mit dem eine selbsterhaltende Kernspaltungs-Kettenreaktion möglich ist. Daher findet es Verwendung als Primärenergieträger in Kernkraftwerken und Kernwaffen.
Geschichte

Uran wurde 1789 von dem deutschen, damals in Berlin lebenden Chemieprofessor und Apotheker Martin Heinrich Klaproth aus dem Mineral Pechblende isoliert. Es ist nach dem Planeten Uranus (und somit nach dem griechischen Himmelsgott Uranos) benannt, der acht Jahre zuvor (1781) von Friedrich Wilhelm Herschel entdeckt worden war. Am 24. September 1789 gab Klaproth die Entdeckung in einer Ansprache vor der Preußischen Akademie der Wissenschaften bekannt. Zuerst wurde seine Entdeckung Uranit genannt, 1790 dann in Uranium umbenannt. Klaproth hatte seine Entdeckung bei der Analyse des Erzes aus dem Bergwerk „Georg Wagsfort“ in Wittigsthal bei Johanngeorgenstadt in Sachsen gemacht. Er behandelte das Erz mit Säure und erwärmte es stark. Das Ergebnis bestand in einem schwarzen Pulver, welches er Uran nannte.
Klaproth hatte tatsächlich ein neues Element identifiziert, aber was er gewonnen hatte, war nicht das Element Uran selbst, sondern ein Oxid. Erst fünfzig Jahre später im Jahre 1841 gelang es dem Franzosen Eugène Peligot, reines Uranmetall zu gewinnen. In der ersten Hälfte des 19. Jahrhunderts wurde Uran zusammen mit anderen Mineralien in St. Joachimsthal sowie in einigen Minen in Cornwall (England) gewonnen.

Uranverbindungen wurden im ganzen 19. Jahrhundert zum Färben von Glas und Keramik verwendet, um Vasen und Dekorationsstücken, aber auch alltäglichen Gebrauchsgegenständen wie Schüsseln, Gläsern etc. eine gelbgrüne Farbe („annagrün“) zu geben. Glashersteller in Joachimsthal (Böhmen) benutzten diese Technik bereits 1826. Noch bis in die Mitte des 20. Jahrhunderts wurde Uran zur Glasfärbung genutzt, erst dann wurde es durch andere, weniger bedenkliche farbgebende Mineralien ersetzt. Uranhaltige keramische Glasuren von Orange bis leuchtend Rot wurden für Geschirr bis hin zu architektonischem Beiwerk verwendet. Diese in den USA aufgrund des Namens eines Herstellers „Fiestaware“ genannte Keramik gehört wohl (neben Americium-Rauchmeldern) zu den radioaktivsten Gegenständen, welche noch immer in vielen amerikanischen Haushalten zu finden sind.
In der Photographie diente bis weit ins 20. Jahrhundert Uranylnitrat zur Braun- und Rottonung von Diapositivplatten, Platinbildern und Bromsilberbildern.[13]
Dass Uran radioaktiv ist, wurde 1896 zuerst von Antoine Henri Becquerel festgestellt.
Uran galt lange als das Element mit der höchsten Ordnungszahl, das natürlich vorkommt. Im Jahr 1971 wurden jedoch winzigste Spuren des Plutoniumisotops 244Pu nachgewiesen, weshalb Plutonium (Z = 94) Uran als natürliches Element mit der höchsten Ordnungszahl ablöste.[14]
Vorkommen

Uran kommt nicht gediegen in der Natur vor, sondern stets in sauerstoffhaltigen Mineralen. Bedeutende Uranminerale sind unter anderem Brannerit und Uraninit (Oxide), Torbernit, Heinrichit und Carnotit (Phosphate, Arsenate und Vanadate) sowie Coffinit und Uranophan (Silikate). Es gibt insgesamt rund 230 Uranminerale, die lokal ebenfalls von wirtschaftlicher Bedeutung sein können. In sedimentären Lagerstätten können sich auch Pseudomorphosen von Uranmineralen (meist Uraninit in Form von Pechblende) nach fossilem Holz oder Bakterien bilden.[15]
Die beiden entscheidenden Faktoren für die Verteilung des radioaktiven Elements Uran auf der Erde sind zum einen der lithophile Charakter des Elements sowie seine unterschiedliche Mobilität in wässrigen Lösungen unter oxidierenden und reduzierenden Bedingungen. Der lithophile Charakter sorgt dafür, dass Uran sich in silikatreichen Schmelzen anreichert. Daher enthalten in der Regel felsische Magmatite wie Granit als Plutonit oder Rhyolith als Vulkanit die höchsten Konzentrationen dieses Elements. Die kontinentale Kruste ist der Bereich der Erde mit den höchsten Urangehalten von durchschnittlich 2,5 ppm, während die ozeanische Kruste und der Erdmantel um Größenordnungen geringere Urangehalte aufweisen. In magmatischen Gesteinen wird Uran meist in akzessorische Minerale wie Zirkon oder Monazit eingebaut, mit welchen man daher sehr gut das Alter der Gesteine datieren kann.
Die unterschiedliche Löslichkeit von Uran unter oxidierenden oder reduzierenden Bedingungen in Lösungen ist der zweite entscheidende Faktor für die Verteilung des Elements und spielt für die Bildung von Uranlagerstätten eine große Rolle. Unter oxidierenden Bedingungen (UO22+) ist Uran in wässrigen Lösungen relativ mobil, während es unter reduzierenden Bedingungen (U4+) schwer löslich ist. Daher sind Redoxgrenzen oftmals lagerstättenkontrollierende Faktoren für das Element.
Ausgehend von oben genannten Faktoren und einigen weiteren gibt es eine große Spannbreite von Uranlagerstätten von magmatischen hydrothermalen bis zu sedimentären Typen. Wichtige Einzeltypen werden von der IAEO unterschieden.
Die höchsten Urangehalte werden in diskordanzgebundenen Lagerstätten mit durchschnittlichen Urangehalten von 0,3 bis 20 % erreicht.[16] Diese stellen derzeit auch die beiden größten Uranproduzenten. Die größte Einzeluranressource der Erde ist Olympic Dam mit einem nachgewiesenen Uraninhalt von über 2 Millionen Tonnen bei durchschnittlichen Urangehalten von etwa 0,03 %.[17] Das erste Uranbergwerk der Welt im industriellen Maßstab in Jáchymov (Tschechische Republik) produzierte aus hydrothermalen Gängen.[18]
Eine Besonderheit stellen die Naturreaktoren von Oklo in Gabun sowie eine benachbarte Uranlagerstätte dar: Von ihnen ist bekannt, dass dort vor etwa 1,5 bis 2 Milliarden Jahren über Jahrtausende Kettenreaktionen in natürlichem Umfeld auftraten, im Zuge derer auch Plutonium-Isotope entstanden. Dies sind weltweit die einzigen Lagerstätten, in welchen das Isotopenverhältnis von Uran-235 zu Uran-238 von den oben angegebenen Daten abweicht, da der Reaktor die Differenz „verbraucht“ hat. Dies wurde entdeckt, als bei der Urananreicherung in Frankreich – trotz der verhältnismäßig kleinen Abweichung – Material für mehrere Atombomben „fehlte“ und deshalb – nicht zuletzt aus Angst vor Proliferation – ermittelt werden musste, wie es dazu kommen konnte. Der Nachweis ungewöhnlicher Isotopenverhältnisse typischer stabiler Spaltprodukte, der nicht anders als durch deren Produktion durch Kernspaltung zu erklären war, lieferte dann den endgültigen Beweis.
Im normalen Boden kommt Uran als Spurenelement vor. Die US-amerikanische Agency for Toxic Substances and Disease Registry (ATSDR) schätzt, dass sich in den obersten 33 cm Erdboden einer Fläche von einer Quadratmeile Land im Mittel ca. 4 Tonnen Uran befinden, also etwa 1,5 Tonnen pro Quadratkilometer.
In Komplexen gebundenes Uran ist auch ein ubiquitäres Element in der Hydrosphäre. Die Urankonzentration in Meerwasser beträgt ca. 3,3 µg/l gegenüber den zum Teil deutlich geringeren Konzentrationen in den Flüssen (0,03 µg/l im Amazonas bis 3,9 µg/l im Ganges). Dies zeigt, dass Uran –wie alle wasserlöslichen Substanzen -– im Meerwasser angereichert wird. Im Meerwasser befindet sich Uran im Gleichgewicht mit der Konzentration in der ozeanischen Kruste, weshalb dessen Entnahme dazu führen würde, dass entsprechende Mengen im Meerwasser gelöst werden, bis sich wieder ein Gleichgewicht bildet. Ähnlich wie bei Vorschlägen Gold aus Meerwasser zu gewinnen, ist jedoch – trotz der weitaus größeren Mengen Uran im Meerwasser im Vergleich zu jener in bekannten Lagerstätten – aufgrund der sehr geringen Konzentration derzeit eine Gewinnung nicht wirtschaftlich. Deutsche Flüsse weisen in der Regel Urankonzentrationen zwischen ca. 1 und 3 µg/l auf. Die Quelle für das Uran liegt in dem geogenen Aufbau der durch die Flüsse entwässerten Gebiete, z. B. können Oberflächenwässer aus Mooren höhere Urankonzentrationen enthalten, und ist somit natürlichen Ursprungs. Lediglich in Ausnahmefällen sind die Urangehalte in Flüssen auf menschlichen Einfluss bspw. die Nutzung uranhaltiger Phosphatdünger und den Uranbergbau (Zwickauer Mulde: ca. 10 µg/l) zurückzuführen. Uran findet sich in Deutschland im unbeeinflussten Grundwasser in Konzentrationen von kleiner 1 bis über 100 µg/l. Die regelmäßige Einnahme von Trinkwasser mit erhöhten Urangehalten kann – aufgrund der chemischen Giftigkeit des Schwermetalls Uran – zum Auftreten von Nierenkrebs führen. Aus diesem Grund empfiehlt die Weltgesundheitsbehörde (WHO) für Trinkwasser einen Grenzwert von 30 µg/l.[19]
Die größten Uranerzreserven liegen nach Angaben der Nuclear Energy Agency (NEA) in den USA, Niger, Australien, Kasachstan, Namibia, Südafrika, Kanada, Brasilien, Russland, Ukraine und Usbekistan.[20] Ehemals bedeutende Lagerstätten im Erzgebirge waren schon zu Zeiten der SDAG Wismut nicht zu Weltmarktpreisen zu gewinnen und sind heute wirtschaftlich nicht mehr interessant.
Uran ist in Spuren auch in Stein- und Braunkohle enthalten. Die weltweit jährlich für die Stromerzeugung verwendete Kohle enthält unter anderem etwa 10.000 t Uran und 25.000 t Thorium, die entweder in die Umwelt gelangen oder sich in Kraftwerksasche und Filterstäuben anreichern. Vereinzelt gibt es daher schon Bestrebungen, Uran aus Kraftwerksasche zu gewinnen.[21] Da trotz Filtern eine gewisser Menge dieser Asche in die Umwelt gelangt, ist die messbare Erhöhung der Radioaktivität im Umfeld von Kohlekraftwerken im Normalbetrieb sogar höher als jene im Umfeld von Kernkraftwerken[22] (wobei hierbei natürlich Störfälle und über das übliche Maß hinausgehende Leckagen nicht berücksichtigt sind).
Der Zusammenhang erhöhter Urangehalte in Mineral- und Trinkwässern mit der Geologie der Grundwasserspeichergesteine wurde 2009 erstmals bundesweit untersucht.[23] Dabei stellte sich heraus, dass erhöhte Urangehalte vorwiegend an Formationen wie Buntsandstein oder Keuper gebunden sind, die selbst geogen erhöhte Urangehalte aufweisen. Allerdings sind örtlich auch bereits Urangehalte aus landwirtschaftlicher Phosphatdüngung in das Grundwasser durchgeschlagen, denn Rohphosphate enthalten 10–200 mg/kg Uran, was bei einer ordnungsgemäßen Düngung zu einem Eintrag von ca. 5 g/ha/a Uran führen kann.
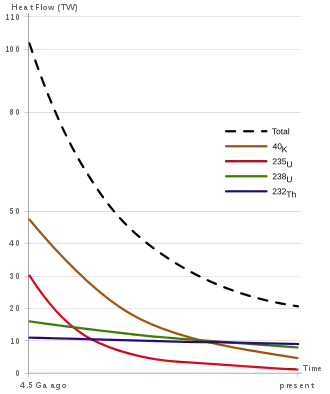
Da Uran eines der schwersten Elemente ist, ist davon auszugehen, dass große Teile des irdischen Urans zu jenen Zeiten, als der gesamte Erdball glutflüssig geschmolzen war, zum Erdkern abgesunken sind. Uran ist neben Thorium und Kalium-40 einer der Hauptbestandteile der Erdwärme welche beständig durch radioaktiven Zerfall „nachgeliefert“ wird.
Abbau
In Deutschland wurde Uran in der Sächsischen Schweiz (Königstein) zuerst konventionell und später durch Laugung, in Dresden (Coschütz/Gittersee insbesondere in Gittersee) und im Erzgebirge (Schlema, Schneeberg, Johanngeorgenstadt, Pöhla) sowie in Ostthüringen (Ronneburg) meist unter Tage als Pechblende durch die SDAG Wismut abgebaut. Geringe Mengen wurden auch im Schwarzwald und im Fichtelgebirge gefördert. Die DDR war damals weltweit der drittgrößte Uranproduzent. Die Abbaugebiete wurden nach 1990 geschlossen, da sie aufgrund des niedrigen Weltmarktpreises unwirtschaftlich waren und der Uranbedarf wegen der geänderten politischen Weltlage (geringere Bedeutung von strategischen Atomwaffen) zurückging. Im Zuge der Sanierung des Standortes Königstein wurde nach 1990 Urankonzentrat als „Nebenprodukt“ der Grubenwasserreinigung auf dem Weltmarkt verkauft. Diese Lieferungen wurden 2021 eingestellt, nachdem auf diesem Weg seit 1990 noch etwa 2.000 Tonnen Urankonzentrat verkauft wurden. Die Einstellung dieser Lieferungen bedeutete gleichzeitig den Ausstieg Deutschlands aus der Reihe uranproduzierender Staaten.[24] Im Gegensatz zu anderen Rohstoffen, deren blasenartige Preisentwicklung Mitte/Ende der 2000er-Dekade zu einem „Berggeschrey“ im Erzgebirge und vereinzelten Versuchen der Wiederaufnahme des Bergbaus führte, gab es, trotz zwischenzeitlich explosionsartig steigenden Uranpreisen (en:Uranium bubble of 2007) zur selben Zeit, keine ernsthaften Versuche, im Erzgebirge wieder im großen Stil Uran abzubauen.
Im Westteil Deutschlands wurden mehrere Kleinst- und Kleinlagerstätten erkundet, jedoch erfolgte einzig in Menzenschwand im Schwarzwald ein nennenswerter Abbau, der 1991 eingestellt wurde. Offiziell diente die Grube Krunkelbach lediglich der „Erkundung“ und angesichts ihrer Stilllegung kurz nach der Wiedervereinigung ist es zumindest denkbar, dass der Abbau des Dual Use-Gutes Uran auch strategischen Zwecken diente, die nach 1990 hinfällig wurden.
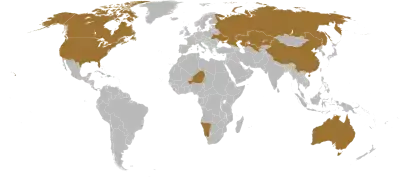 Die zehn Staaten mit der weltweit größten Uranförderung (2008)
Die zehn Staaten mit der weltweit größten Uranförderung (2008)
Die Weltproduktion von Uran betrug im Jahr 2006 39.603 Tonnen. Große Förderländer sind Australien, Kanada, Russland, Niger, Namibia, Kasachstan, Usbekistan, Südafrika und die USA. Der Verbrauch lag 2006 weltweit bei 66.500 Tonnen und wird von der Internationalen Atomenergieorganisation (IAEO) durch den Neubau von Kernkraftwerken für das Jahr 2030 auf 93.775 bis 121.955 Tonnen geschätzt. Der Abbau deckt etwa 60 % des aktuellen Bedarfs, der Rest wird durch Lagerbestände, Wiederaufarbeitung und abgerüstete Kernwaffen gedeckt.[25] Schätzungen der IAEO, Greenpeace und der Atomwirtschaft über die Reichweite der Uran-Vorkommen liegen unterschiedliche Angaben über die weltweiten Ressourcen und den zukünftigen Verbrauch zugrunde. Sie liegen zwischen 20 und 200 Jahren.[26]
Durch den Uranbergbau werden Uran und radioaktive Zerfallsprodukte (z. B. das radioaktive Edelgas Radon) aus dem Untergrund an die Oberfläche verbracht. Die damit verbundene Freisetzung führt zu Schäden an Umwelt und Gesundheit.[27]
Darstellung
Verarbeitung von Uranerz
Uranerze, z. B. Uraninit (Pechblende, U3O8) oder Carnotit (KUO2VO4 · 1,5 H2O), werden sauer mit Schwefelsäure oder auch alkalisch mit Soda aufgeschlossen.
Die nach dem sauren Aufschluss entstandenen Lösungen werden mit Ammoniak behandelt, worauf der Yellow Cake ausfällt. Dieser enthält hauptsächlich Ammoniumdiuranat ((NH4)2U2O7) und noch weitere Polyuranate, Uranylhydroxide und -sulfate. Die Lösung des alkalischen Aufschlusses wird mit NaOH versetzt, wodurch Natriumdiuranat (Na2U2O7) ausfällt. Um das Natrium zu entfernen, wird es dann in H2SO4 gelöst und anschließend mit wässrigem NH3 als (NH4)2U2O7 ausgefällt.
Der „Yellow Cake“ wird in Salpetersäure (HNO3) gelöst, wobei unlösliche Anteile ausfallen und durch Filtration oder Zentrifugieren entfernt werden. Aus der Lösung kann dann rohes Uranylnitrat (UO2(NO3)2) auskristallisiert werden. Eine Lösung von Uranylnitrat wird dann mit Tributylphosphat (TBP) extrahiert (PUREX-Prozess), nach Eindampfen und Waschen wird reines Uranylnitrat erhalten.
Vorsichtige Pyrolyse führt zu den verschiedenen Modifikationen von Uran(VI)-oxid (UO3), je nach Temperatur und Sauerstoffdruck.[28][29][30] Zur Gewichtsreduktion beim Transport wird der „Yellow Cake“ thermisch zersetzt, worauf schwarzes U3O8 entsteht.
 Pulverförmiger Yellowcake
Pulverförmiger Yellowcake


Reindarstellung
Uran(VI)-oxid (UO3) wird mit Wasserstoff zu Uran(IV)-oxid (UO2) reduziert.[31] Bringt man Urandioxid mit wasserfreiem Fluorwasserstoff zur Reaktion, so entsteht Urantetrafluorid, aus welchem schließlich durch Reduktion mittels Calcium oder Magnesium reines Uran gewonnen wird[32]:

Uran kann generell durch die Reduktion von Uranhalogeniden mit Alkali- oder Erdalkalimetallen hergestellt werden[32]:


Ebenso kann auch eine Elektrolyse von KUF5 oder UF4 in geschmolzenem Calciumchlorid (CaCl2) / Natriumchlorid (NaCl) erfolgen. Sehr reines Uran kann durch die thermische Zersetzung von Uranhalogeniden an einem Glühdraht erzeugt werden.[33] Aus Urandioxid ist es u. a. durch Reduktion mit Calcium erhältlich.[34]

Eigenschaften

Physikalische Eigenschaften
Uran ist ein relativ weiches, silber-weißes Metall hoher Dichte, welches in drei Modifikationen vorkommt.[1][35]
| Phase | stabiler Temperaturbereich |
Dichte (Temp.) |
Kristallsystem (Achsen in Pikometer) |
|---|---|---|---|
| α-Uran | unterhalb 688 °C | orthorhombisch (a = 285,4, b = 586,9, c = 495,6) | |
| β-Uran | 688 °C bis 776 °C | tetragonal (a = 1075,9, c = 565,6) | |
| γ-Uran | oberhalb 776 °C | kubisch (a = 352,5) |
Uran-Rhodium-Germanium (URhGe) ist die erste entdeckte Legierung, die in sehr starken Magnetfeldern eine eintrittsinvariante Supraleitung zeigt.[36]
Chemische Eigenschaften
Uran ist in fein verteiltem Zustand selbstentzündlich. Die meisten Säuren lösen metallisches Uran auf, während es von Basen nicht angegriffen wird. An der Luft überzieht sich das Metall mit einer Oxidschicht.
Uran bildet eine Reihe von Verbindungen, in denen es in den Oxidationsstufen +2 bis +6 vorliegen kann. Die Farbe von Urankomplexen ist in der Regel stark von der Oxidationszahl, aber auch von der Ligandenumgebung abhängig. In wässriger Lösung, ebenso wie in festen Verbindungen werden häufig die folgenden Kombinationen von Farbe und Oxidationsstufe beobachtet: U3+ (violett), U4+ (grün), UVO2+ (blasslila) und UVIO22+ (gelb).[37] In nichtwässrigen Lösungen mit organischen Liganden ergeben sich häufig andere Farbkombinationen. Uran tritt in der Natur überwiegend mit den Wertigkeiten +4 oder +6 auf. Vierwertige Uranminerale sind in Wasser unter normalen pH/EH-Bedingungen nahezu unlöslich. Uranverbindungen sind giftig. Die Toxizität wird v. a. durch deren Löslichkeit bestimmt. Die leichtlöslichen Uranyl-Salze sind am giftigsten, die schwerlöslichen Oxide sind weniger giftig. Uran ist teratogen.
Biologische Aspekte
Bei der Gattung Desulfovibrio wurde die Fähigkeit, Uran als Elektronenakzeptor zu verwenden, nachgewiesen: Uran(VI) wird zu Uran(IV) reduziert. Desulfovibrio vulgaris verwendet Cytochrom-c3 als Uran-Reduktase.[38] Wenn Uran(VI) als einziger für das Bakterium nutzbarer Elektronenakzeptor vorliegt, wurde allerdings kein Wachstum beobachtet.[39] Ein Bakterium, welches Uran(VI) als einzigen Elektronenakzeptor nutzen kann und dabei auch Wachstum zeigt, ist Geobacter metallireducens der Geobacteraceae.[40]
Unlösliches Uran kann durch bakterielle Aktivität mobilisiert werden. Unter aeroben Bedingungen können die Eisen-Schwefel-Bakterien Acidithiobacillus ferrooxidans und Leptospirillum ferrooxidans Pyrit (FeS2) zu Eisen(II)-sulfat (FeSO4) und dann zu Eisen(III)-sulfat (Fe2(SO4)3) oxidieren. Eisen(III)-Ionen können unlösliches Uran(IV) zu löslichem Uran(VI) oxidieren.[41]
Die Reduktion von löslichem Uran(VI) zu unlöslichem Uran(IV) durch Prokaryoten wurde als mögliche Methode zur biologischen Sanierung von Uran-kontaminierten Grundwässern und gefährlichen Abfällen vorgeschlagen.[42][43]
Isotope
Von Uran sind 25 Isotope und 3 Kernisomere mit Halbwertszeiten zwischen 1 µs und 4,468 Milliarden Jahren bekannt.[44] Nur die vier langlebigsten Isotope kommen in der Natur vor. Davon stammen 238U und 235U noch aus der Entstehungszeit des Sonnensystems, sie wurden im r-Prozess in Supernovae gebildet. 234U entsteht über mehrere Zwischenstufen beim Zerfall aus 238U, 236U durch seltene Neutroneneinfänge aus 235U. Das künstlich erzeugbare fünftlanglebigste Isotop 233U spielt in der Technik ebenfalls eine Rolle. Alle anderen Isotope haben Halbwertszeiten von maximal 68,9 Jahren.
In natürlichem Uran (Natururan) finden sich deshalb die Isotope 238U zu 99,27 %, 235U zu 0,72 %, 234U zu 0,0055 % und 236U in Spuren. Das Isotopenverhältnis der Uranisotope ändert sich im Laufe der Zeit, da 238U und 235U unterschiedlich schnell zerfallen. Die Häufigkeit des dritten natürlichen Isotops 234U bleibt im Verhältnis zur Häufigkeit des 238U konstant, da 234U ein Zerfallsprodukt des 238U ist und mit diesem im Gleichgewicht steht.
Ein anderes Verhältnis der Uranisotope findet sich im Bereich der Naturreaktoren, von denen Oklo in Gabun der zuerst entdeckte und der bekannteste ist. Bei dem heutigen Isotopenverhältnis von 235U und 238U ist das Auftreten einer derartigen natürlichen Reaktorzone nicht mehr möglich.
- 238U hat eine Halbwertszeit von 4,468 Milliarden Jahren und ist wie die anderen natürlichen Isotope (234U und 235U) ein α-Strahler. Die spezifische Aktivität von 238U beträgt 12.450 Bq/g. 238U ist der natürliche Beginn der Uran-Radium-Reihe.
- 235U hat eine Halbwertszeit von 703,8 Mio. Jahren. Es ist der natürliche Beginn der Uran-Actinium-Reihe. Es ist spaltbar und hat einen Anteil von etwa 0,7 % in natürlichem Uranvorkommen. Aufgrund seiner Spaltbarkeit hat es große wirtschaftliche Bedeutung.
- 234U hat eine Halbwertszeit von 245.500 Jahren. Es ist wegen seiner relativ kurzen Halbwertszeit im Vergleich zu 238U nur in Spuren vorhanden, liefert aber einen gleich großen Beitrag zur Radioaktivität wie letzteres. Es entsteht gemäß:
- Die Zeitangaben sind Halbwertszeiten.

- 236U ist ein α-Strahler mit einer Halbwertszeit von 23,42 Millionen Jahren und kommt in der Natur nur in Spuren vor.[45] Es entsteht durch Neutroneneinfang aus 235U. Wenn Uran einem erhöhten Neutronenfluss ausgesetzt ist, wie z. B. in einem Kernreaktor, erhöht sich der Anteil an 236U deutlich.[46] Die Anteile der Isotope 234U, 235U, 236U in einer Urankontamination können Aufschluss über deren Ursprung geben.[47] 236U zerfällt über die bis zum natürlichen Plutonium 244Pu verlängerte Thorium-Reihe.
- 233U hat eine Halbwertszeit von 159.200 Jahren und ist spaltbar. Es ist nicht im natürlichen Uran enthalten, sondern wird in Brutreaktoren wie dem THTR-300 aus dem schwer spaltbaren Thorium 232Th (Spalt-Wirkungsquerschnitt 3 µb, wie beim 238U) erbrütet. 233U zerfällt über die Neptunium-Reihe.
- Die Zeitangaben sind Halbwertszeiten.

Spaltbarkeit

(Zum Abspielen Großansicht öffnen)
{kind=link}
Der Wirkungsquerschnitt für induzierte Kernspaltung durch ein thermisches Neutron ist bei 233U und 235U mit 530 bzw. 586 b (Barn) groß,[48] bei 238U dagegen mit nur 3 µb sehr klein. Im technisch-praktischen Sinn sind also nur die Isotope 233 und 235 „gut spaltbar“ und damit mögliche Brennstoffe für Kernreaktoren.
Als „angereichert“ wird Uran bezeichnet, dessen Anteil an 235U gegenüber dem 238U durch Uran-Anreicherung erhöht wurde. Schwach angereichertes Uran (im Fachjargon „LEU“, lightly enriched uranium) wird in Kernkraftwerken, hochangereichertes Uran („HEU“ highly enriched uranium) für Forschungszwecke, in der Medizin[49], in den Reaktoren der US Navy[50] und zur Herstellung von Kernwaffen verwendet. Die Grenze zwischen LEU und HEU wird gewöhnlich bei einem Anreicherungsgrad des 235U von 20 % festgesetzt.
Die kritische Masse von 235U beträgt etwa 49 kg; hier ist der Wirkungsquerschnitt der schnellen Spaltung ausschlaggebend, da ein Moderator fehlt. Mit einem 20 cm dicken Wasserreflektor lässt sich die kritische Masse auf 22 kg, mit einem 30 cm-Stahlreflektor auf 16,8 kg absenken. In wässriger Lösung lässt sich die kritische Masse bei einer optimalen Dichte von 0,059 g/cm³ mit Reflektor auf unter 600 g verringern.[51]
Die kritische Masse von 233U beträgt nur rund 16 kg. Auch hier lässt sich mit einem Reflektor die kritische Masse absenken: etwa 7,4 kg mit 20 cm Wasser und 6,2 kg mit 30 cm Stahl. In wässriger Lösung lässt sich die kritische Masse auf 425 g verringern. Das Isotop kann in Kernreaktoren aus 232Th durch Neutroneneinfang und zwei anschließende Betazerfälle erbrütet werden.
Das Isotop mit der geringsten kritischen Masse (3,6 kg) ist 232U. Es ist wie alle geradzahligen Uranisotope beinahe ausschließlich durch schnelle Neutronen spaltbar. Mit Stahlreflektor kann die kritische Masse auf 1,9 kg reduziert werden. Das Isotop ist jedoch nur sehr aufwändig zu gewinnen, da es sich nicht in einem Kernreaktor herstellen lässt.
Das vierte Uran-Isotop, das eine Kettenreaktion aufrechterhalten kann, ist 234U. Seine kritische Masse beträgt 145 kg. Es ist als Folgeprodukt von 238U im natürlichen Uran enthalten und kann auch aus 233U oder 232Th erbrütet werden. Wegen der hohen kritischen Masse und der im Vergleich zu anderen spaltbaren Isotopen umständlichen Gewinnung spielt es in der Kerntechnik keine größere Rolle. Es wird allerdings bei den meisten Anreicherungsmethoden relativ gesehen noch stärker angereichert als 235U, was wegen der geringen Ausgangskonzentration aber meist nicht ins Gewicht fällt.
Die Isotope 236U und 238U können selbst keine Kettenreaktion tragen. 238U wird jedoch in Verbindung mit anderen, spaltbaren Isotopen in einem schnellen Brüter als Brutstoff eingesetzt. Durch Neutroneneinfang und anschließende Betazerfälle entsteht dann 239Pu, das wiederum gut spaltbar ist.
Verwendung und Verbreitung
Zivile Nutzungen
Das Uranisotop 235U wird in Kernkraftwerken zur Energiegewinnung genutzt. Das Isotop 238U kann in Brutreaktoren eingesetzt werden, um Plutonium herzustellen. Das Isotop 235U kommt in nur geringer Konzentration (etwa 0,72 %) im natürlichen Uran vor und wird durch Anreicherung konzentriert. Die zurückbleibende Fraktion wird abgereichertes Uran genannt (Abreicherung).
Bei der Spaltung eines 235U-Atomkerns werden durchschnittlich 210 MeV Energie freigesetzt. Davon sind etwa 190 MeV in einem Reaktor thermisch verwertbar.[52] Die Spaltung von 1 g 235U liefert daher etwa 0,95 MWd (Megawatt-Tage) = 22,8 MWh thermische Energie.
Ein Kilogramm 235U hat einen als Wärme nutzbaren Energiegehalt von 76 Terajoule bzw. 2,5 Mio. Steinkohleeinheiten.[53]
Die aus 1 kg Natururan tatsächlich erzeugte Strommenge hängt vom eingesetzten Reaktortyp und dem Brennstoffkreislauf ab und liegt etwa bei 36–56 MWh für den Fall der direkten Endlagerung der abgebrannten Brennelemente, also ohne Wiederaufarbeitung und ohne Brüten.[25] (Nicht berücksichtigt ist der Energieaufwand für Bergbau, evtl. Anreicherung, Transport und Endlagerung.)
Aufgrund seiner hohen Absorptionswirkung für ionisierende Strahlung wird abgereichertes Uran (depleted uranium, DU) im Strahlenschutz als Abschirmmaterial eingesetzt. DU wird aufgrund seiner hohen Dichte für Trimmgewichte in Flugzeugen und Kielgewichte für Hochleistungssegelboote verwendet. Nach der Diskussion, die durch den Absturz einer Frachtmaschine in Amsterdam ausgelöst worden war, wird es in Flugzeugen durch Wolfram ersetzt.[54]
Vor allem in der ersten Hälfte des 20. Jahrhunderts erfreute sich Uran auch als Bestandteil von Vaselineglas sowie Uranglasuren für Keramik einer großen Beliebtheit in den USA.[55]
Militärische Nutzungen
Kernreaktoren werden zum Antrieb großer Kriegsschiffe eingesetzt, etwa für Flugzeugträger und U-Boote. Allerdings besitzen nur wenige Länder nuklear angetriebene Kriegsschiffe. Jeder der 10 Flugzeugträger der Nimitz-Klasse der US-Navy besitzt 2 Reaktoren mit jeweils 140 MW Leistung.
235U ist neben Plutonium das wichtigste Ausgangsmaterial für den Bau von Kernwaffen und Zündsätzen für Wasserstoffbomben.
Viele Streitkräfte nutzen Uranmunition.[56] Das ist panzerbrechende Munition, die abgereichertes Uran als Projektilkernmaterial enthält. Die Munition wird beim Eintritt in den Panzerinnenraum zerstäubt und verbrennt dabei explosionsartig. Die entstehenden Stäube und Aerosole sind giftig und führen bei kontaminierten Personen zu Gesundheitsschäden.[57] Urangeschosse sind keine Hartkerngeschosse, sondern gehören zur allgemeineren Übergruppe der Wuchtgeschosse. Uran ist im Vergleich zu Wolfram zwar von ebenso großer Dichte, jedoch weniger hart und schmilzt früher. Uran steht Atomwaffenstaaten als Abfallprodukt aus der Anreicherung zur Verfügung. Die Beschaffung und Verarbeitung sind billiger als die der Alternative Wolfram.
In jüngster Zeit wurden in indisch-pakistanischen Grenzkonflikten, in Tschetschenien, während der sowjetischen Invasion Afghanistans, im Kosovo-Krieg, im Zweiten Golfkrieg und in der Militärinvasion der USA und anderer Staaten in den Irak (Dritter Golfkrieg) von den beteiligten Parteien insgesamt mehrere Hundert Tonnen Uranmunition eingesetzt.
Abgereichertes Uran wird bei Panzern – wie dem M1 Abrams – auch als Panzerung eingesetzt. Es handelt sich um eine Sandwichpanzerung mit einer Schicht Uran zwischen zwei Schichten Panzerstahl. Eine Folge der militärischen Verwendung ist die legale wie illegale Verbreitung uran- und auch plutoniumhaltigen Materials.
Verbindungen
→ Kategorie: Uranverbindung
Oxidationsstufen

Uran bildet eine Reihe von Verbindungen, in denen es in den Oxidationsstufen +2 bis +6 vorliegen kann. Es tritt in der Natur überwiegend mit den Wertigkeiten +4 oder +6 auf.
Uran (II)
Im Jahre 2013 wurde erstmals Uran in der Oxidationsstufe +2 dargestellt. Die Synthese gelang durch Reduktion einer Tris(cyclopentadienyl)-Uran(III)-Verbindung unter Verwendung in situ erzeugter Alkalide.[58] Die Existenz molekularer Uran(II)-Verbindungen konnte auch durch die Reduktion eines Tris(aryloxid)aren-Uran(III)-Komplexes verifiziert werden, welcher chemisch durch elementares Kalium in Gegenwart von 2.2.2-Kryptand,[59] sowie elektrochemisch bei einem Potential von -2,50 V (gegen Fc+/Fc) reduziert werden konnte.[60] Die beiden Uran(II)-Komplexe besitzen eine unterschiedliche Elektronenkonfiguration, welche durch das Ligandenfeld bestimmt wird. Der Tris(cyclopentadienyl)-Uran(II)-Komplex besitzt die Elektronenkonfiguration [Rn] 5f36d1,[58] wohingegen die Tris(aryloxid)aren-Uran(II)-Verbindung eine [Rn]-5f4-Konfiguration annimmt.[59]
Uran (III)
Die erste Uran(III)-Verbindung wurde 1842 von Peligot als UCl3 dargestellt. Das U3+-Ion ist ein starkes Reduktionsmittel (Reduktionspotential zwischen -2,2 und 1,5 V gegen Fc+/Fc) und setzt in Gegenwart von Wasser Wasserstoff frei.[61] Uran(III)-Verbindungen sind in sauerstoff- und halogenfreien organischen Lösungsmitteln relativ stabil und unter anaeroben Bedingungen haltbar. Die Synthese von niedervalenten U(III) Komplexen erfolgt in der Regel ausgehend von UI3, oder dem daraus hergestellten UHMDS. UI3 wird hierzu aus metallischem Uran mit elementarem Iod synthetisiert. Das besondere Interesse an niedervalentem Uran ergibt sich aus der hohen Reaktivität gegenüber kleinen Molekülen mit biologischer und industrieller Relevanz, wie z. B. CO, CO2, N2, NO, oder H2O.[62]
Uran (IV)
Von den Verbindungen des Uran(IV) sind vor allem das Oxid (UO2) und die vier Halogenide (UF4, UCl4, UBr4 und UI4) bekannt. Es handelt sich um Feststoffe mit hohen Schmelzpunkten über 500 °C.
Uran (V)
Seit 2003 sind Uranyl(V)-Verbindungen im Festkörper bekannt.[63] Seither wurden eine Vielzahl weiterer Uranyl(V)-Verbindungen synthetisiert.[64]
Uran (VI)
Das sechswertige Uran kommt in der Natur ausschließlich in Form der UO22+-Gruppe (Uranyl-Gruppe) vor, d. h., es gibt kein U6+. Diese Gruppe bindet sich bevorzugt an sauerstoffhaltige Bindungspartner: als Phosphat, Sulfat, Carbonat und mit Wasser als Hydroxid. Uranylacetat und Uranylnitrat sind lösliche Uransalze. Diese Salze sind kommerziell erhältlich und entsprechen in ihrer Giftigkeit anderen Schwermetallnitraten und -acetaten.
 Uranylacetat
Uranylacetat
(UO2(CH3COO)2) Uranylnitrat
Uranylnitrat
(UO2(NO3)2)
Oxide
→ Übersicht: Uranoxide
Urandioxid (UO2) ist ein schwarzes, kristallines Pulver, das im späten 19. Jahrhundert bis in die Mitte des 20. Jahrhunderts als Keramikglasur verwendet wurde. Heutzutage wird es vor allem als nuklearer Brennstoff in Brennstäben eingesetzt. Bekannt sind auch Urantrioxid (UO3), Triuranoctoxid (U3O8) und Uranylperoxid (UO2O2).
Yellowcake

Yellowcake ist ein Uranoxidkonzentrat. Der Name ist abgeleitet von der früheren Farbe und Textur. Heutzutage werden höhere Kalzinationstemperaturen verwendet, wodurch der „gelbe Kuchen“ eher dunkelgrün bis schwarz ist. Ursprünglich waren die im Yellowcake enthaltenen chemischen Verbindungen unbekannt. Man nahm an, dass es sich um Ammoniumdiuranat oder Natriumdiuranat handelt. Die Zusammensetzung variierte und hing vom Verhüttungsprozess ab. Die Verbindungen, die in Yellowcake gefunden wurden, sind unter anderem Uranylhydroxid, Uranylsulfat, Natrium-para-Uranat und Uranylperoxid, zusammen mit einer Reihe von Uranoxiden. Der heutige Yellowcake enthält typischerweise zu 70 bis 90 Prozent (Massenanteil) das Uranoxid (U3O8).
Das hellgelbe Ammoniumdiuranat ist ein Zwischenprodukt bei der Herstellung von Yellowcake. Manchmal wird es ebenfalls als „Yellowcake“ bezeichnet, das entspricht allerdings nicht dem allgemeinen Gebrauch.
Halogenide

Für Uran sind Halogenide in den Oxidationsstufen +3 bis +6 bekannt. Für die Stufen +3 bis +5 sind sämtliche Verbindungen der vier Halogene Fluor, Chlor, Brom und Iod bekannt, für die Oxidationsstufe +6 sind es UF6 und UCl6.[65]
| Oxidationszahl | F | Cl | Br | I |
|---|---|---|---|---|
| +6 | Uran(VI)-fluorid UF6 farblos |
Uran(VI)-chlorid UCl6 grün |
||
| +5 | Uran(V)-fluorid UF5 farblos |
Uran(V)-chlorid UCl5 braun |
Uran(V)-bromid UBr5 schwarz |
(Uran(V)-iodid) (UI5) |
| +4 | Uran(IV)-fluorid UF4 grün |
Uran(IV)-chlorid UCl4 grün |
Uran(IV)-bromid UBr4 braun |
Uran(IV)-iodid UI4 schwarz |
| +3 | Uran(III)-fluorid UF3 purpur |
Uran(III)-chlorid UCl3 rot |
Uran(III)-bromid UBr3 rot |
Uran(III)-iodid UI3 schwarz |

Urantetrafluorid (UF4), auch bekannt als „green salt“, ist ein Zwischenprodukt der Herstellung von Uranhexafluorid.
Uranhexafluorid (UF6) ist ein weißer Feststoff, der bei einer Temperatur von 56,5 °C sublimiert und nur unter Druck von mind. 1,5 bar und 64,1 °C eine flüssige Phase bildet. UF6 ist die Uranverbindung, die für die zwei häufigsten Anreicherungsprozesse, Gasdiffusion und Gaszentrifuge, verwendet wird. Es wird in der Industrie schlicht als „Hexe“ bezeichnet.
Metallorganische Verbindungen
Uranocen U(C8H8)2 ist eine der ersten Organouranverbindungen und die bekannteste Verbindung des Cyclooctatetraen mit den f-Elementen.[66][67] Weiterhin sind beispielsweise zu nennen auch das an der Luft stabile Derivat U(C8H4Ph4)2 und das Cycloheptatrienylion [U(C7H7)2]−.[68]
Analytik
Klassische qualitative Analytik von Uran
Uran tritt in Lösung meist als UO22+-Kation auf. Im anorganischen Trennungsgang wird UO22+ in der Ammoniumsulfid-Urotropin-Gruppe nachgewiesen. Nach zahlreichen Trennungs- und Fällungsschritten wird es als UO2(SCN)2·3 Ether in eine Etherphase extrahiert.
Der Nachweis erfolgt durch Zugabe von gelbem Blutlaugensalz (K4[Fe(CN)6]), wobei sich bei relativ hohen Konzentrationen ein Mischkomplex bildet (K2(UO2[Fe(CN)6])). Dieser fällt als rotbrauner Niederschlag aus.[69]
Thermisch angeregte optische Spektroskopie
Atomabsorptionsspektrometrie (AAS) in Form der Flammen-AAS und Induktiv-gekoppeltes-Plasma optische Emissionsspektrometrie ICP-OES werden auf Grund der geringen Empfindlichkeit nur in seltenen Ausnahmen für die Analytik eingesetzt. Wegen Störungen durch starke Carbidbildung wird auch die Graphitrohr-AAS nur selten eingesetzt.
Induktiv-gekoppelte Plasma-Massenspektrometrie (ICP-MS)
Mittels ICP-MS wird Uran sehr empfindlich gemessen, die in der Natur vorkommenden drei Uranisotope können direkt bestimmt werden. So kann z. B. das Isotop 238U (99,274 %) in menschlichen Haarproben mittels ICP-MS mit einer Nachweisgrenze von 0,2 ng/g bestimmt werden.[70]
Neutronenaktivierungsanalyse (NAA)
In der NAA wird bei der Bestimmung von Uran die Aktivierungsreaktion 238U(n,γ)239U genutzt. 239U besitzt eine Halbwertszeit von 23,5 min. Zur quantitativen Auswertung wird der Photopeak mit einer Gammastrahlungsenergie von 74 keV herangezogen. Mit dieser hochempfindlichen Methode wurde eine Nachweisgrenze von 4 pg/ml Uran in Meerwasser erzielt.[71]
Fluoreszenzspektrometrie
Uran in Form von Uranylionen (UO22+) kann mit Hilfe der zeitaufgelösten, laserinduzierten Fluoreszenzspektrometrie (TR-LIF) quantitativ bestimmt werden. Diese Methode findet häufig Anwendung in der Überwachung und zum Nachweis von Uran in Grund- und Oberflächengewässern in der Nähe von Aufbereitungsanlagen, da sie die Möglichkeit der on-line Überwachung bietet. Nachweisgrenzen von 40 ng/l wurden mit dieser Technik erzielt.[72]
Adsorptive kathodische Stripping-Voltammetrie (AdCSV)
Für die Uranbestimmung in Ab-, Grund- und Trinkwässern wird zunehmend die AdCSV verwendet. Das UO22+-Ion wird dabei in saurem Milieu mit Chloranilsäure komplexiert und elektrochemisch bei 150 mV an einer hängenden Quecksilbertropfelektrode (HMDE) adsorptiv angereichert. Anschließend wird ein Voltammogramm von 50 mV bis −200 mV aufgenommen. Der Reduktionspeak liegt etwa bei −90 mV. Mit diesem Verfahren wurde eine Nachweisgrenze von 24 ng/l erzielt. Aufgrund der sehr guten Selektivität wurde das Verfahren als Standardverfahren (DIN 38406-17) genormt.[73]
Sicherheitshinweise
Uran ist aufgrund seiner Radioaktivität gefährlich und, wie die meisten Schwermetalle, chemisch giftig.
Chemisch gefährlich sind vor allem wasserlösliche Uranverbindungen, welche analog zu Blei, Cadmium und Quecksilber vorwiegend die Tubuli der Nieren schädigen.[74][75] Die Weltgesundheitsorganisation WHO empfahl 2003 – angesichts der Verwendung von abgereichertem Uran in Uranmunition – einen Grenzwert für die tägliche Aufnahme von löslichen Uranverbindungen von 0,5 μg/kg Körpergewicht, von 5 μg/kg für unlösliche Verbindungen und von maximal 1 μg/m³ in der Umgebungsluft bei Aufnahme über den Atemtrakt.[76] Bei oraler Aufnahme von Uran und -verbindungen werden dabei zwischen 0,2 und 2 %, beim Einatmen etwa 5 % resorbiert, der Rest über den Harn ausgeschieden.[76]
Seine Radioaktivität erzeugt Ionisierende Strahlung, welche Auslöser von Erbgutveränderungen – wie Mutationen – und nachfolgenden Krebserkrankungen sein kann. Die langlebigen Uranisotope sind α-Strahler, die im Fall einer Aufnahme in den Körper eine relativ hohe Strahlendosis zur Folge haben. Beim Umgang und Lagerung von Uran und seinen Verbindungen ist zu beachten, dass aus den Uran-Zerfallsreihen Folgeprodukte anwesend sind, die auch Beta- und durchdringende Gammastrahlen emittieren; daneben auch Radon, das als Gas seinen Weg überallhin findet.
Literatur
- Ingmar Grenthe, Janusz Drożdżyński, Takeo Fujino, Edgar C. Buck, Thomas E. Albrecht-Schmitt, Stephen F. Wolf: Uranium. In: Lester R. Morss, Norman M. Edelstein, Jean Fuger (Hrsg.): The Chemistry of the Actinide and Transactinide Elements. Springer, Dordrecht 2006, ISBN 1-4020-3555-1, S. 253–698 (doi:10.1007/1-4020-3598-5 5).
- Walter D. Loveland, David Morrissey, Glenn T. Seaborg: Modern Nuclear Chemistry. Wiley-Interscience, 2006, ISBN 0-471-11532-0.
- Robert J Schwankner, Gerolf Lieckfeld, Doris Lienert: Die Frühgeschichte des Urans. In: Die Geowissenschaften. 7, 8, 1989, S. 215–224 (doi:10.2312/geowissenschaften.1989.7.215).
Weblinks
- Eintrag zu Uran. In: Römpp Online. Georg Thieme Verlag, abgerufen am 3. Januar 2015.
- Uranium: Human Health Fact Sheet (Memento vom 6. Februar 2004 im Internet Archive) (engl.) (PDF-Datei; 46 kB)
- Peter Eichstaedt: Uranium, Chemical & Engineering News, 2003
- Uran – Informationen des Bundesamts für Lebensmittelsicherheit und Veterinärwesen
Einzelnachweise
- Harry H. Binder: Lexikon der chemischen Elemente. S. Hirzel Verlag, Stuttgart 1999, ISBN 3-7776-0736-3, S. 674–682.
- Die Werte der atomaren und physikalischen Eigenschaften (Infobox) sind, wenn nicht anders angegeben, aus www.webelements.com (Uranium) entnommen.
- CIAAW, Standard Atomic Weights Revised 2013.
- Eintrag zu uranium in Kramida, A., Ralchenko, Yu., Reader, J. und NIST ASD Team (2019): NIST Atomic Spectra Database (ver. 5.7.1). Hrsg.: NIST, Gaithersburg, MD. doi:10.18434/T4W30F (https://physics.nist.gov/asd). Abgerufen am 13. Juni 2020.
- Eintrag zu uranium bei WebElements, https://www.webelements.com, abgerufen am 13. Juni 2020.
- Die Werte der atomaren und physikalischen Eigenschaften (Infobox) sind, wenn nicht anders angegeben, aus www.webelements.com (Uranium) entnommen.
- A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 102. Auflage. Walter de Gruyter, Berlin 2007, ISBN 978-3-11-017770-1, S. 2149.
- Robert C. Weast (Hrsg.): CRC Handbook of Chemistry and Physics. CRC (Chemical Rubber Publishing Company), Boca Raton 1990, ISBN 0-8493-0470-9, S. E-129 bis E-145. Werte dort sind auf g/mol bezogen und in cgs-Einheiten angegeben. Der hier angegebene Wert ist der daraus berechnete maßeinheitslose SI-Wert.
- Die Werte der atomaren und physikalischen Eigenschaften (Infobox) sind, wenn nicht anders angegeben, aus www.webelements.com (Uranium) entnommen.
- Eintrag zu Uranium im Classification and Labelling Inventory der Europäischen Chemikalienagentur (ECHA), abgerufen am 1. August 2016. Hersteller bzw. Inverkehrbringer können die harmonisierte Einstufung und Kennzeichnung erweitern.
- Eintrag zu Uran in der GESTIS-Stoffdatenbank des IFA, abgerufen am 9. August 2016. (JavaScript erforderlich)
- Die von der Radioaktivität ausgehenden Gefahren gehören nicht zu den einzustufenden Eigenschaften nach der GHS-Kennzeichnung.
- Fritz Schmidt: Kompendium der praktischen Photographie. 10., wesentlich verbesserte Auflage. Leipzig 1906. Urannitrat S. 191, Urantonung S. 268, 291, 339.
- D. C. Hoffman, F. O. Lawrence, J. L. Mewherter, F. M. Rourke: Detection of Plutonium-244 in Nature. In: Nature. Band 234, 1971, S. 132–134 (doi:10.1038/234132a0).
- Helmut Tonndorf: Metallogenie des Urans im ostdeutschen Zechstein – Ein Beitrag zum Nachweis und zur Charakteristik stofflicher Umverteilungs- und Anreicherungsprozesse. (= Abhandlungen der sächsischen Akademie der Wissenschaften zu Leipzig, Mathematisch-naturwissenschaftliche Klasse. Band 58, Heft 3). Akademie Verlag, Berlin 1994, ISBN 3-05-501621-1.
- Geology of Uranium deposits.
- K. Ehrig, BHP Billiton. Präsentation auf der South Australia Explorers Conference, Adelaide 2008.
- Verschiedene Autoren: Uranbergbau im Erzgebirge und Kalter Krieg. Ausgewählte Beiträge des RADIZ-Workshops vom 10. und 11. Oktober 1997 in Schlema, RADIZ-Information 16/1998, RADIZ e.V., Schlema.
- Broder Merkel, Britta Planer-Friedrich, Christian Wolkersdorfer (Hrsg.): Uranium in the Aquatic Environment. Springer-Verlag, Heidelberg 2002, ISBN 3-540-43927-7.
- Uranium 2020: Resources, Production and Demand. NEA, abgerufen am 26. Oktober 2021 (englisch).
- World Nuclear Association: Naturally-Occurring Radioactive Materials (NORM); März 2009.
- https://www.scientificamerican.com/article/coal-ash-is-more-radioactive-than-nuclear-waste/
- F. Knolle: Ein Beitrag zu Vorkommen und Herkunft von Uran in deutschen Mineral- und Leitungswässern. TU Braunschweig, 2009 (PDF)
- Letzte Uran-Laster starten in Königstein In: Sächsische Zeitung (Ausgabe Pirna) vom 1. Juni 2021.
- OECD Nuclear Energy Agency und Internationale Atomenergieorganisation: Uranium 2007: Resources, Production and Demand. OECD Publishing, 2008, ISBN 978-92-64-04768-6 (englisch, online [PDF; abgerufen am 7. Juli 2009]).
- Uran ist ein begehrter Rohstoff geworden. In: FAZ. 6. Januar 2006.
- Der schmutzigste Atombrennstoff, Fragen und Antworten zur Herkunft des Urans (PDF; 790 kB (Memento vom 30. Januar 2012 im Internet Archive)).
- Federation of American Scientists: Uranium Production.
- Frank Settle: Nuclear Chemistry, Uranium Production. (Memento vom 25. Juni 2013 im Webarchiv archive.today)
- Carl Willis: Uranium Chemistry.
- A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 102. Auflage. Walter de Gruyter, Berlin 2007, ISBN 978-3-11-017770-1, S. 1950.
- Georg Brauer: Uran. In: Handbuch der Präparativen Anorganischen Chemie. Ferdinand Enke Verlag, Stuttgart 1954, S. 1069–1073.
- C. R. Hammond: The Elements. In: Handbook of Chemistry and Physics. 81. Auflage. CRC Press, 2000, ISBN 0-8493-0481-4 (online [PDF]).
- Wilhelm Jander: Über Darstellung von reinem Uran. In: Zeitschrift für anorganische und allgemeine Chemie. 138 (1), 1924, S. 321–329 (doi:10.1002/zaac.19241380131).
- Georg Brauer (Hrsg.): Handbuch der Präparativen Anorganischen Chemie. 3., umgearb. Auflage. Band II. Enke, Stuttgart 1978, ISBN 3-432-87813-3, S. 1195.
- F. Levy, I. Sheikin, B. Grenier, A. D. Huxley: Magnetic Field-Induced Superconductivity in the Ferromagnet URhGe. In: Science. 309, 2005, S. 1343–1346 (doi:10.1126/science.1115498).
- A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 102. Auflage. Walter de Gruyter, Berlin 2007, ISBN 978-3-11-017770-1, S. 1956.
- D. R. Lovley, P. K. Widman, J. C. Woodward, E. J. Phillips: Reduction of Uranium by Cytochrome c3 of Desulfovibrio vulgaris. In: Appl. Environ. Microbiol. 59 (11), 1993, S. 3572–3576. PMID 8285665; PMC 182500 (freier Volltext).
- D. R. Lovley, E. J. Phillips: Reduction of Uranium by Desulfovibrio desulfuricans. In: Appl. Environ. Microbiol. 58 (3), 1992, S. 850–856. PMID 1575486; PMC 195344 (freier Volltext).
- D. R. Lovley, E. J. P. Phillips, Y. A. Gorby, E. R. Landa: Microbial Reduction of Uranium. In: Nature. 350, 1991, S. 413–416 (doi:10.1038/350413a0).
- O. H. Tuovinen, T. M. Bhatti: Microbiological Leaching of Uranium Ores. In: Minerals and Metallurgical Processing. 16, 1999, S. 51–60; doi:10.1007/BF03403234.
- D. R. Lovley: Bioremediation of Organic and Metal Contaminants with Dissimilatory Metal Reduction. In: J. Ind. Microbiol. 14 (2), 1995, S. 85–93. PMID 7766214.
- The Biochemical Periodic Tables – Uranium.
- Nuclear Wallet Card – Z(92) (engl.)
- P. Steier, R. Golser, W. Kutschera, V. Liechtenstein, A. Priller, A. Valenta, C. Vockenhuber: Heavy ion AMS with a small accelerator, Nuclear Instruments and Methods. In: Physics Research B. 188 (1–4), 2002, S. 283–287 (doi:10.1016/S0168-583X(01)01114-4).
- M. A. C. Hotchkis, D. Child, D. Fink, G. E. Jacobsen, P. J. Lee, N. Mino, A. M. Smith, C. Tuniz: Measurement of 236U in environmental media, Nuclear Instruments and Methods. In: Physics Research B. 172 (1–4), 2000, S. 659–665 (doi:10.1016/S0168-583X(00)00146-4).
- D. L. Donohue: Strengthening IAEA safeguards through environmental sampling and analysis. In: Journal of Alloys and Compounds. 271–273, 1998, S. 12–18 (doi:10.1016/S0925-8388(98)00015-2).
- G. Pfennig, H. Klewe-Nebenius, W. Seelmann-Eggebert (Hrsg.): Karlsruher Nuklidkarte. 7. Auflage. 2006.
- world-nuclear.org: Radioisotopes in Medicine (Memento vom 26. Dezember 2013 im Internet Archive).
- Ending the Production of Highly Enriched Uranium for Naval Reactors. (PDF 235 kB, S. 2(87)) cns.miis.edu, abgerufen am 12. April 2016 (englisch).
- Institut de Radioprotection et de Sûreté Nucléaire: Evaluation of nuclear criticality safety data and limits for actinides in transport. S. 16 (PDF (Memento vom 18. November 2014 im Internet Archive)).
- Kernenergie – woher kommt sie? Zukunftswerkstatt Jena
- https://www.kernd.de/kernd-wAssets/docs/service/018basiswissen.pdf Martin Volkmer: Kernenergie Basiswissen, Mitteilung Deutsches Atomforum e.V., Seite 35, abgerufen am 26. Juni 2021
- U.S. Nuclear Regulatory Commission: Systematic Radiological Assessment of Exemption for Source and Byproduct Materials. Abschnitt 3.17, S. 531–533 (PDF; 3,3 MB).
- Daniel Rhodes: Clay and Glazes for the Potter. 2015.
- Uni Oldenburg: Informationen über Uran-Munition.
- E. S. Craft, A. W. Abu-Qare, M. M. Flaherty, M. C. Garofolo, H. L. Rincavage, M. B. Abou-Donia: Depleted and natural Uranium: Chemistry and toxicological effects. In: Journal of Toxicology and Environmental Health – Part B – Critical Reviews. 7 (4), 2004, S. 297–317 (doi:10.1080/10937400490452714).
- Matthew R. MacDonald, Megan E. Fieser, Jefferson E. Bates, Joseph W. Ziller, Filipp Furche, William J. Evans: Identification of the +2 Oxidation State for Uranium in a Crystalline Molecular Complex, [K(2.2.2-Cryptand)][(C5H4SiMe3)3U]. In: J. Am. Chem. Soc. 135, 2013, S. 13310–13313 (doi:10.1021/ja406791t).
- Henry S. La Pierre, Andreas Scheurer, Frank W. Heinemann, Wolfgang Hieringer, Karsten Meyer: Synthesis and Characterization of a Uranium(II) Monoarene Complex Supported by δ Backbonding. In: Angewandte Chemie International Edition. 53 (28), 2014, S. 7158–7162 (doi:10.1002/anie.201402050).
- Henry S. La Pierre, Hajime Kameo, Dominik P. Halter, Frank W. Heinemann, Karsten Meyer: Coordination and Redox Isomerization in the Reduction of a Uranium(III) Monoarene Complex. In: Angewandte Chemie International Edition. Band 53, Nr. 28, 7. Juli 2014, S. 7154–7157, doi:10.1002/anie.201402048.
- William J. Evans, Stosh A. Kozimor: Expanding the chemistry of U3+ reducing agents. In: Coordination Chemistry Reviews (= Actinide Chemistry). Band 250, Nr. 7, 1. April 2006, S. 911–935, doi:10.1016/j.ccr.2006.01.017.
- Wiley: Progress in Inorganic Chemistry, Volume 58 – Kenneth D. Karlin. Abgerufen am 14. Oktober 2017.
- J.-C. Berthet, M. Nielich, M. Ephritikhine: Isolation of a Uranyl [UO2]+ Species: Crystallographic Comparison of the Dioxouranium(V) and (VI) Compounds [UO2(OPPh3)4](OTf)n (n=1, 2). In: Angew. Chem. Int. Ed. 42, 2003, S. 1952 (doi:10.1002/anie.200250506).
- Polly L. Arnold, Jason B. Love, Dipti Patel: Pentavalent uranyl complexes. In: Coordination Chemistry Reviews. 253 (15–16), 2009, S. 1973–1978 (doi:10.1016/j.ccr.2009.03.014).
- A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 102. Auflage. Walter de Gruyter, Berlin 2007, ISBN 978-3-11-017770-1, S. 1969.
- A. Streitwieser, U. Müller-Westerhoff: Bis(cyclooctatetraenyl)uranium (Uranocene). A New Class of Sandwich Complexes That Utilize Atomic f Orbitals. In: J. Am. Chem. Soc. 90 (26), 1968, S. 7364–7364 (doi:10.1021/ja01028a044).
- Christoph Elschenbroich: Organometallchemie. 6. Auflage. Wiesbaden 2008, ISBN 978-3-8351-0167-8, S. 587–591.
- D. Seyferth: Uranocene. The First Member of a New Class of Organometallic Derivatives of the f Elements. In: Organometallics. 23 (15), 2004, S. 3562–3583 (doi:10.1021/om0400705).
- G. Jander, E. Blasius: Lehrbuch der analytischen und präparativen anorganischen Chemie. 16. Auflage. S. Hirzel Verlag, Stuttgart 2006, ISBN 3-7776-1388-6.
- H. Sela, Z. Karpas, C. Pickhardt, J.S. Becker: Biomonitoring of hair samples by laser ablation inductively couple plasma mass spectrometry (LA-ICP-MS). In: International Journal of Mass Spectrometry. 261 (2–3), 2007, S. 199–207 (doi:10.1016/j.ijms.2006.09.018).
- R. Lobinski, Z. Marczenko: Spectrochemical Trace Analysis for Metals and Metalloids. Elsevier, Amsterdam 1997.
- G. Romanovskaya, V. Pogonin, A. Chibisov: Determination of Trace Amounts of Uranium(VI) in Various Materials by a Repetitive Laser Technique. In: Talanta. 34 (1), 1987, S. 207–210 (doi:10.1016/0039-9140(87)80028-0; PMID 18964281).
- G. Henze: Polarographie – Voltammetrie; Grundlagen und analytische Praxis. Springer Verlag, Berlin/ Heidelberg/ New York 2001.
- Thomas Efferth: Molekulare Pharmakologie und Toxikologie: Biologische Grundlagen von Arzneimitteln und Giften. Springer, 2006, ISBN 3-540-21223-X, S. 238.
- Werner Böcker, Helmut Denk, Philipp Ulrich Heitz: Repetitorium Pathologie. Elsevier, Urban & Fischer, 2007, ISBN 978-3-437-43400-6, S. 296.
- WHO: Depleted uranium: sources, exposure and health effects. Executive summary, Januar 2003 (PDF; 23 kB).