Nukleophile Substitution
Die nukleophile Substitution ist ein Reaktionstyp in der organischen Chemie. Hierbei reagiert ein Nukleophil in Form einer Lewis-Base (Elektronenpaardonator) mit einer organischen Verbindung vom Typ R–X (R bezeichnet einen Alkyl- oder Arylrest, X ein elektronenziehendes Heteroatom). Das Heteroatom wird dabei durch das Nukleophil ersetzt (siehe Substitutionsreaktion).
Allgemeines Beispiel zur nukleophilen Substitution, in dem X für ein Halogen – wie Chlor, Brom oder Iod – steht:
| Nukleophile Substitution | ||||
n-Butanol | ||||
| Halogenalkan + Nukleophil –––––> Substitutionsprodukt + Halogenid | ||||
In der Anorganischen Chemie ist dieser Typus ebenfalls anzutreffen, ein Beispiel ist die Hydrolyse von Siliciumtetrachlorid.
Allgemeine Kennzeichen
Nukleophile Substitutionsreaktionen werden meistens in Lösung durchgeführt. Dabei sind die Polarität des Lösungsmittels sowie die Substituenteneinflüsse in den Edukten von entscheidender Bedeutung für die Geschwindigkeit der Reaktion. Ist das Lösungsmittel selbst der nukleophile Reaktionspartner, spricht man von einer Solvolyse.[1]
Edukte
Nukleophile
Als Nukleophile können die verschiedensten Verbindungen eingesetzt werden. Dabei handelt es sich um Anionen oder elektronenreiche Moleküle mit freien Elektronenpaaren (siehe Beispiele unten).
Typ R-X
Das angegriffene Molekül R-X hat eine stark polare Bindung (ungleiche Verteilung der Elektronendichte), z. B. C-Cl, C-Br, C-O, C=O oder Si-Cl.
In folgenden Verbindungen kann das Heteroatom beziehungsweise die Heteroatom-haltige Gruppe durch ein Nukleophil substituiert werden:
- Alkylhalogenide: Alkylchloride und Alkylbromide[2]
- Arylhalogenide: Arylchloride und Arylbromide
- Carbonsäurederivate, wie -Chloride, Ester, und -Anhydride
- Sulfonsäureester, z. B. Tosylate (p-Toluolsulfonsäureester) oder Mesylate (Methansulfonsäureester) oder die besonders reaktiven Triflate (Trifluormethansulfonsäureester)
- Oxiran-, Thiiran- und Aziridin-Ringe (→ Heterocyclen)
Mechanismen
Nukleophile Substitutionen werden bei aliphatischen und aromatischen Verbindungen beobachtet: Es gibt aliphatische nukleophile Substitutionen und aromatische nukleophile Substitutionen, wobei erstere wesentlich weiter verbreitet ist.
Darüber hinaus werden die Reaktionen aufgrund der Molekularität in verschiedene Gruppen eingeteilt. Das heißt, die Reaktionen werden danach eingeordnet, wie viele Moleküle am geschwindigkeitsbestimmenden Schritt der Reaktion beteiligt sind. Die beschriebenen Mechanismen SN1 und SN2 sind als Extremfälle der nukleophilen Substitution aufzufassen. Der Übergang dazwischen ist fließend. Eine Zusammenfassung beider Reaktionen findet sich in der folgenden Tabelle:
| Zusammenfassung der SN1- und SN2-Reaktion | ||
|---|---|---|
| Reaktion | SN1 | SN2 |
| Kinetik | v = k·c[Substrat] | v = k·c[Substrat]·c[Nukleophil] |
| primäres Alkyl | niemals, außer bei stabilisierenden Gruppen | exzellent, außer bei sterisch gehindertem Nukleophil |
| sekundäres Alkyl | moderat | moderat |
| tertiäres Alkyl | sehr gut | niemals |
| Benzyl | exzellent | moderat, außer bei tertiärem Benzyl |
| Allyl | exzellent | moderat, außer bei tertiärem Allyl |
| Abgangsgruppe | wichtig | wichtig |
| Nukleophilie | unwichtig | wichtig |
| Bevorzugtes Lösungsmittel | polar protisch | polar aprotisch |
| Stereochemie | Racemisierung bei Substitution am Stereozentrum | Inversion (Chemie) bei Substitution am Stereozentrum durch Walden-Umkehr |
| Umlagerung | häufig | selten |
| Eliminierung | meist, besonders bei basischem Nukleophil | nur mit Hitze und basischem Nukleophil |
Der SNi-Mechanismus ist ein Spezialfall, der gesondert diskutiert wird.
Aromatische nukleophile Substitutionen laufen meistens zweistufig ab, das heißt die Zwischenprodukte sind oft isolierbar (siehe Meisenheimer-Komplexe). Zusätzlich ist ein sogenannter Dehydrobenzol-Mechanismus bekannt, der auch als Arinmechanismus bezeichnet wird.
SN1-Mechanismus
Bei der SN1-Reaktion steht „SN“ für eine nukleophile Substitution und die „1“ für einen „monomolekularen“ Mechanismus, der zwar zweistufig verläuft, aber bei dem nur ein Molekül am geschwindigkeitsbestimmenden Schritt beteiligt ist.
Kinetik der SN1-Reaktion
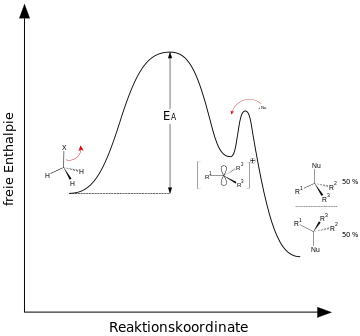
Aus dem Geschwindigkeitsgesetz einer Reaktion kann versucht werden, auf den Reaktionsmechanismus zu schließen. Bei der SN1-Reaktion findet man ausschließlich eine Abhängigkeit von der Konzentration des Substrats. Die Reaktionsgeschwindigkeit v errechnet sich somit mit Hilfe des Geschwindigkeitsgesetzes aus einer Geschwindigkeitskonstante k sowie der Konzentration des Substrats c[Substrat]:

Die Reaktionsgeschwindigkeit ist in erster Ordnung von dem Substrat abhängig und in nullter Ordnung von dem Nukleophil (also gar nicht). Die Gesamtordnung der Reaktion beträgt also eins. Der Reaktionsmechanismus besteht zwar aus zwei Schritten, die Reaktionsgeschwindigkeit ist jedoch nur vom langsamsten Reaktionsschritt abhängig; dieser ist der geschwindigkeitsbestimmende Schritt. Der geschwindigkeitsbestimmende Schritt kann mit einem Flaschenhals verglichen werden: Die Geschwindigkeit, mit der Wasser in eine Flasche strömen kann, wird nur von der engsten Stelle der Flasche kontrolliert – dem Flaschenhals.
Mechanismus
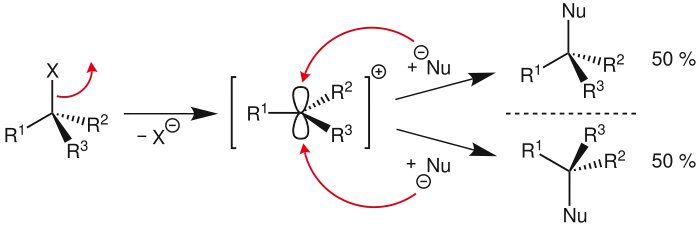
Die SN1-Reaktion verläuft in zwei Schritten. Im ersten, geschwindigkeitsbestimmenden (= langsamsten) Schritt dissoziiert die Verbindung R-X und setzt die Abgangsgruppe X− als Anion frei. Zurück bleibt ein planares, sp2-hybridisiertes Carbokation (Carbeniumion) R+. Dieses reaktive Zwischenprodukt (R+) wird im zweiten Schritt von dem Nukleophil Nu angegriffen. Dieser zweite Schritt ist im Verhältnis zum ersten sehr schnell. Das Nukleophil ist am geschwindigkeitsbestimmenden Schritt in Gegensatz zur SN2-Reaktion nicht beteiligt. Außerdem wird der SN1-Mechanismus durch eine relativ kleine Anfangskonzentration der Edukte begünstigt. Stehen aber mehrere Nukleophile zur Verfügung, so findet man im Produkt überwiegend das stärkere Nukleophil wieder.
Stereochemie
Der geschwindigkeitsbestimmende Schritt beim SN1-Mechanismus ist die Bildung des idealerweise planaren Carbokations. Die Konfiguration der Ausgangsverbindung wird dadurch aufgehoben.
Theoretisch ist der nachfolgende Angriff des Nukleophils von beiden Seiten gleich wahrscheinlich. Ein racemisches Produkt wäre die Folge, da der Angriff von der der austretenden Gruppe gegenüberliegenden Seite einen Konfigurationswechsel (Inversion), der von derselben Seite die Erhaltung der Konfiguration (Retention) zur Folge hätte. Experimentell findet man oft mehr Produkte mit einem Konfigurationswechsel (Ausbeute 50–70 %); dies liegt daran, dass nach einer Abspaltung der Abgangsgruppe diese Abgangsgruppe nicht schnell genug durch das Lösungsmittel diffundiert und somit diese Angriffsrichtung versperrt. Die abgehende Gruppe kann sich vor dem nukleophilen Angriff nicht ausreichend entfernen und stellt so für das angreifende Nukleophil eine Behinderung dar. Dies führt manchmal zu einer verstärkten Inversion der Konfiguration. In einem solchen Fall spricht man von einer Teil-Racemisierung.
Beobachtet wurden bei Reaktionen nach SN1 jedoch alle stereochemischen Möglichkeiten von vollständiger Inversion bis zur Racemisierung, wobei die Bildung eines Racemates die Regel ist.
Einflussfaktoren
- Polarität des Lösungsmittels: Je polarer das Lösungsmittel ist, desto besser kann es die SN1-Reaktion durch Wasserstoffbrückenbindungen stabilisieren und beschleunigt dadurch die Reaktion. Besonders deutlich tritt dieser Effekt in einem protischen Lösungsmittel zutage. Die SN1-Reaktion profitiert von solchen Lösungsmitteln, da sowohl Übergangszustand wie auch Zwischenprodukt polar bzw. geladen sind. Der Übergangszustand des geschwindigkeitsbestimmenden Schritts der SN1-Reaktion ist zunächst durch Ladungstrennung polar: Die negativ geladene Abgangsgruppe entfernt sich und lässt ein positiv geladenes Carbokation zurück. Das entstehende Zwischenprodukt ist eine geladene Verbindung (das Carbokation), außerdem wird ein negativ geladenes Nukleophil freigesetzt. Bei der SN2-Reaktion hingegen ist der Übergangszustand nicht polar, da Ladung nur verschoben, aber nicht erzeugt wird.
Es kann also gesagt werden, dass die SN1-Reaktion von protischen Lösungsmitteln bevorzugt wird.
- Güte der Abgangsgruppe: Da im geschwindigkeitsbestimmenden Schritt die Abgangsgruppe das Molekül verlässt, wird die Reaktionsgeschwindigkeit stark von ihrer Qualität beeinflusst. Die Abgangsgruppe ist häufig negativ geladen. Die Leichtigkeit, mit der sie das Molekül verlässt, hängt mit ihrer Fähigkeit zusammen, diese negative Ladung zu stabilisieren. Diese Fähigkeit, Ladung zu stabilisieren, ist umso stärker, je weniger basisch das Nukleophil X− ist bzw. je stärker sauer die konjugierte Säure HX ist. (Nukleofugie)
- Substratstruktur: Je höher ein Kohlenstoffatom substituiert ist, desto schneller läuft die SN1-Reaktion an ihm ab. Ein tertiäres Alkyl reagiert also schneller als ein sekundäres Alkyl und dieses schneller als ein primäres Alkyl, dies liegt an der Stabilität der Carbokationen. Diese Stabilität hängt mit Hyperkonjugation und dem +I-Effekt zusammen: Alkylgruppen liefern dem Carbokation als Substituenten Elektronendichte und verringern auf diese Weise die positive Ladung. So wird einerseits das Carbokation stabilisiert und dissoziiert zudem leichter. Primäre Alkyle sind so instabil, dass sie keine SN1-Reaktion mehr eingehen.
Ein weiterer Faktor ist, dass mit zunehmender Substitution sterische Spannung verringert wird.
Auch Allylreste wirken mesomeriestabilisierend und stabilisieren somit das Carbenium-Ion, sowie auch Benzylsubstituenten.
- Carbokationumlagerung: Ein Carbokation kann sich durch einen Hydrid- oder Methyl-Shift umlagern, wenn so eine stabilere Verbindung entsteht. Aus einem sekundären kann beispielsweise ein tertiäres Carbokation entstehen. So können unterschiedliche Produkte entstehen, wenn SN1- und SN2-Reaktion am selben Molekül ablaufen.
SN2-Mechanismus
SN2 steht für eine nukleophile Substitution mit einem bimolekularen Mechanismus, der einstufig verläuft und bei dem beide Moleküle am geschwindigkeitsbestimmenden Schritt beteiligt sind.
Kinetik von SN2-Reaktionen

Einblicke in den Reaktionsmechanismus bietet das Geschwindigkeitsgesetz. Bei kinetischen Untersuchungen findet man eine Abhängigkeit der Reaktionsgeschwindigkeit v von der Konzentration des Substrats c[Substrat] und von der Konzentration des Nukleophils c[Nukleophil], was zusammen mit einer Geschwindigkeitskonstante k zu folgendem Geschwindigkeitsgesetz führt:

Das Geschwindigkeitsgesetz erklärt sich durch den Reaktionsmechanismus, der nur aus einem einzigen Schritt besteht – dieser ist natürlich zugleich der geschwindigkeitsbestimmende Schritt. Bei diesem tritt zeitlich mit dem Angriff des Nukleophils die Abgangsgruppe aus. Die Reaktionsgeschwindigkeit steigt ebenso proportional mit einer zunehmenden Zahl an angreifenden Molekülen (steigender Konzentration des Nukleophils) wie mit einer zunehmenden Zahl an angegriffenen Molekülen (steigender Substrat-Konzentration), da beides die Wahrscheinlichkeit für einen erfolgreichen Zusammenstoß erhöht. Da bei der SN2-Reaktion am geschwindigkeitsbestimmenden Schritt zwei Moleküle beteiligt sind, handelt es sich um eine Reaktion zweiter Ordnung. Bei der SN2-Reaktion steht das „S“ für Substitution, das „N“ für nukleophil und die „2“ für die Bimolekularität bzw. die 2. Ordnung.[3] Da bei der SN2-Reaktion Bindungsbildung und -bruch gleichzeitig stattfinden, handelt es sich um eine konzertierte Reaktion. Dies bedeutet gleichzeitig, dass die Reaktion ohne nachweisbare Zwischenprodukte abläuft und nur einen Übergangszustand besitzt.[4]
Mechanismus
Die SN2-Reaktion verläuft immer über einen Rückseitenangriff; das bedeutet, dass das Nukleophil von der gegenüberliegenden Seite angreift, an der die Abgangsgruppe gebunden ist. Dies lässt sich damit erklären, dass das angreifende Nukleophil der ebenfalls negativ geladenen Abgangsgruppe im Weg stünde, falls beide auf derselben Seite wären. Für eine leistungsfähigere Erklärung dagegen ist die Molekülorbitaltheorie notwendig:
Um eine chemische Bindung auszubilden, muss das HOMO des einen Moleküls mit dem LUMO des anderen Moleküls in Wechselwirkung treten. Bei der SN2-Reaktion muss das besetzte nichtbindende Molekülorbital (HOMO) des Nukleophils mit dem unbesetzten, antibindenden Molekülorbital (LUMO) der Kohlenstoffverbindung eine Wechselwirkung eingehen. Bei einem Rückseitenangriff kommt es zu bindender Wechselwirkung, bei einem Vorderseitenangriff jedoch zu bindender und antibindender Wechselwirkung gleichzeitig. Daher erfolgt der erfolgreiche Angriff eines Nukleophils immer von der Rückseite her. Aus der Notwendigkeit eines Rückseitenangriffs heraus erklärt sich auch, dass bei zunehmender Methylierung eines Alkans (dem Ersetzen von Wasserstoffatomen durch Methylgruppen) die Reaktionsgeschwindigkeit fortlaufend abnimmt.
Vor der Reaktion liegt das Kohlenstoffatom sp3-hybridisiert vor, also tetraedrisch. Während der Reaktion nähert sich das Nukleophil dem positiv polarisierten Kohlenstoffkern; im Übergangszustand bildet sich eine trigonale Bipyramide mit schwach gebundenen axialen Liganden. Damit ist gemeint, dass die Bindungselektronenpaare der drei Reste, die nicht an der eigentlichen Reaktion beteiligt sind, in dieselbe Ebene rücken und sich das Nukleophil sowie die Abgangsgruppe auf ihrer jeweiligen Seite wie die Spitzen einer Pyramide auf einer Achse senkrecht zur beschriebenen Ebene gegenüberstehen. Die ganze Reaktion ist als ein fließender Übergang zu verstehen. Die Bindungen zwischen Kohlenstoff und Nukleophil sowie Kohlenstoff und Abgangsgruppe sind jeweils geschwächt, da es sich um eine 3-Zentren-4-Elektronen-Bindung handelt.
Aus diesem Mechanismus resultiert eine Inversion der Konfiguration des Kohlenstoffatoms; diese wird Walden-Umkehr oder auch "Regenschirmprinzip nach Krieger" genannt, da die tetraedrische Anordnung des Kohlenstoffs an einen Regenschirm erinnert, der im Verlauf der Reaktion wie durch einen Windstoß umgestülpt wird. Diese Inversion spielt nur bei chiralen Molekülen eine Rolle. Durch diese Inversion würde somit aus einer nach der Cahn-Ingold-Prelog-Nomenklatur benannte (S)- Verbindung eine (R)-Verbindung.[3] Die Inversion kann genutzt werden, um ein bestimmtes Enantiomer gezielt zu synthetisieren. Soll die Konfiguration erhalten bleiben, können zwei aufeinanderfolgende SN2-Reaktionen durchgeführt werden; dies führt zu einer Retention (Erhaltung) der Konfiguration.[4]
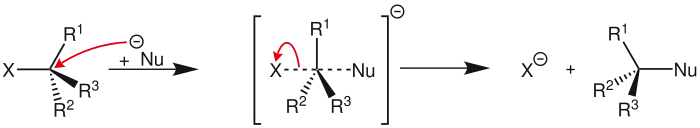 Sn2-Mechanismus
Sn2-Mechanismus- Dargestellt ist der Verlauf der SN2-Reaktion. Auf der linken Seite ist das Kohlenstoffatom ebenso wie auf der rechten Seite tetraedrisch, in der Mitte fünfbindig trigonal-bipyramidal, die Reste R1 bis R3 befinden sich alle axial in einer Ebene.
Außerdem wird der Reaktionsverlauf durch eine relativ große Anfangskonzentration beider Edukte begünstigt. Wasser hingegen unterdrückt die SN2-Reaktion.[5]
Stereochemie
Die Walden-Umkehr führt zur Inversion am stereochemischen Zentrum.
Einflussfaktoren
- Polarität des Lösungsmittels: Je besser ein Nukleophil solvatisiert ist, desto schlechter ist es. Die Wahl des Lösungsmittels sollte demnach ausfallen. Der SN2-Mechanismus findet bevorzugt in polaren aprotischen Lösungsmitteln statt.
- Abgangsgruppe/ Nukleofug: siehe SN1
- Nukleophilie: Die Güte eines Nukleophils nennt sich Nukleophilie. Die Nukleophilie hängt von der Ladung, der Basizität und der Polarisierbarkeit des Nukleophils, dem Lösungsmittel und den Substituenten ab. Zusätzliche negative Ladung verstärkt die Nukleophilie. Damit ist eine Base immer nukleophiler als ihre konjugierte Säure. Dies liegt daran, dass die bei einem Angriff des Nukleophils sich ausbildende Bindung zu dem elektrophilen Kohlenstoffatom umso leichter bilden kann, je elektronenreicher das Nukleophil ist. Die stärkere Base ist auch das stärkere Nukleophil. Dadurch nimmt die Nukleophilie im Periodensystem von links nach rechts ab. Allerdings kommt diesem Trend eine geringere Bedeutung zu als der Anwesenheit einer Ladung.[4]
- Substratstruktur: Je höher ein Kohlenstoffatom substituiert ist, desto langsamer läuft die SN2-Reaktion an ihm ab. So ist die Reaktionsgeschwindigkeit v von:SN2-Reaktionen an einem tertiären Kohlenstoffatom laufen faktisch nicht ab, sondern werden durch Konkurrenz von Nebenreaktionen verdrängt (wie der SN1-Reaktion). Die Abnahme der Reaktionsgeschwindigkeit entsteht durch den Raumbedarf der Methylgruppen. Da eine Methylgruppe ein größeres Volumen einnimmt, als ein Wasserstoffatom, blockiert sie den möglichen Angriff des Nukleophils – dies nennt sich sterische Hinderung. Die sterische Hinderung nimmt nicht nur mit der Anzahl, sondern auch mit der Länge der Alkanreste zu – je länger sie sind, desto stärker behindern sie eine mögliche Reaktion bzw. desto stärker senken sie die Reaktivität des Moleküls in einer SN2-Reaktion.Verzweigungen am Cα-Kohlenstoff erhöhen den sterischen Anspruch und hindern die Reaktion somit noch stärker.

Konkurrenz zwischen SN1- und SN2-Reaktion
Die SN1- und die SN2-Reaktion stehen in Konkurrenz zueinander. Bei der Synthese einer Verbindung würde man eine SN2-Reaktion einer SN1-Reaktion vorziehen, da die SN2-Reaktion zu einem einzigen Produkt, die SN1-Reaktion jedoch zu einem Gemisch aus wenigstens zwei Produkten führt und durch Carbokationenumlagerung weiter verkompliziert wird. Somit wird bei einer Synthese versucht, die Bedingungen für eine SN2-Reaktion herzustellen. Ob eine Reaktion eher nach einem SN1- oder SN2-Mechanismus abläuft, wird von folgenden Faktoren beeinflusst:
- der Struktur der Verbindung
- der Konzentration des Nukleophils
- der Reaktivität des Nukleophils
- dem Lösungsmittel
Bei der Struktur der Verbindung ist zunächst entscheidend, ob das Kohlenstoffatom, das die zu substituierende Gruppe trägt, ein primäres, sekundäres oder tertiäres Kohlenstoffatom ist. Der erste, davon abhängige Effekt, ist, dass mit zunehmendem Alkylierungsgrad das bei einer SN1-Reaktion entstehende Carbokation durch I-Effekte und Hyperkonjugation zunehmend besser stabilisiert werden kann. So läuft z. B. eine SN1-Reaktion an tert-Butylbromid leichter ab als an 2-Brompropan.
Der zweite Effekt ergibt sich, wie bereits erwähnt, durch sterische Hinderung. Da die SN2-Reaktion über einen Rückseitenangriff abläuft und dieser durch den Raumbedarf von Alkylgruppen erschwert wird, wird eine SN2-Reaktion mit zunehmender Alkylierung schwieriger. So läuft z. B. eine SN2-Reaktion an 2-Brompropan leichter ab als an tert-Butylbromid. Somit wird mit zunehmender Alkylierung eine SN2-Reaktion erschwert, eine SN1-Reaktion hingegen erleichtert.
Die Abhängigkeit der SN1- und SN2-Reaktion von Konzentration und Reaktivität des Nukleophils lassen sich durch eine Betrachtung des Geschwindigkeitsgesetzes verstehen. So lautet das Geschwindigkeitsgesetz für eine SN2-Reaktion:

Eine SN2-Reaktion ist also von der Konzentration des Eduktes und der Konzentration des Nukleophils abhängig. Das Geschwindigkeitsgesetz für eine SN1-Reaktion lautet:
Die Reaktionsgeschwindigkeit ist also nur von der Konzentration des reagierenden Moleküls abhängig. Wenn, wie im Falle einer Konkurrenzsituation, beide Reaktionen ablaufen können, so lautet das Geschwindigkeitsgesetz der Gesamtreaktion:

An den Geschwindigkeitsgesetzen ist zu erkennen, dass die Konzentration des Nukleophils einen Einfluss auf SN2-Reaktionen besitzt, nicht jedoch auf SN1-Reaktionen. Somit folgt, dass in einer Konkurrenzsituation zwischen SN1- und SN2-Reaktion die SN2-Reaktion durch eine Erhöhung der Konzentration des Nukleophils begünstigt werden kann.
Eine weitere Möglichkeit zur Bevorzugung der SN2-Reaktion liegt in der Veränderung der Güte (Reaktivität) des Nukleophils. Eine SN2-Reaktion besteht nur aus einem einzigen Schritt: Das Nukleophil verdrängt die Abgangsgruppe. Eine SN1-Reaktion besteht dagegen aus dem ersten, langsamen Schritt (der damit geschwindigkeitsbestimmend ist), bei dem sich die Abgangsgruppe vom Molekül löst und einem zweiten, schnellen Schritt, bei dem das Nukleophil rasch mit dem resultierenden Molekül reagiert. Ein gutes Nukleophil wird somit eine SN2-Reaktion beschleunigen, eine SN1-Reaktion dagegen nicht.[3]
Die Stabilität des entstehenden Kations wird von seinem Lösungsmittel beeinflusst. So können Kationen und Anionen in polaren Lösungsmitteln durch Solvatation stabilisiert werden. So kann die Nucleophilie durch Wasserstoffbrückenbindungen erniedrigt werden, daher sind Nucleophile in aprotischen Lösungsmitteln reaktiver und SN2-Reaktionen verlaufen schneller, wohingegen SN1-Reaktionen eher in protischen Lösungsmitteln stattfinden.[6]
SN2-Mechanismen am ungesättigten Kohlenstoffatom
Betrachtet man chlorsubstituierte ungesättigte Verbindungen, wie Vinylchlorid (C2H3Cl) oder Chlorbenzol (C6H5Cl), so wird gefunden, dass diese ungesättigten Verbindungen nur äußerst schlecht von Nukleophilen wie dem Hydroxid-Ion oder dem Amid-Ion angegriffen werden. Alkylhalogenide, also die gesättigten Halogenverbindungen, reagieren meist bereits bei Raumtemperatur, während bei der Reaktion von Chlorbenzol mit Hydroxid-Ionen Temperaturen von 200 °C nötig sind. Verantwortlich für dieses reaktionsträge Verhalten ist die erhöhte Elektronendichte an den ungesättigten Kohlenstoffatomen. Dadurch wird der Angriff eines Nukleophils erschwert; ungesättigte Kohlenstoffatome ziehen das gemeinsame Elektronenpaar der zu substituierenden Gruppe (z. B. die C-Cl-Bindung im Vinylchlorid oder im Chlorbenzol) stärker zu sich, was das Abstrahieren des Chloratoms erschwert.
Die Einführung elektronenziehender Gruppen im Benzol und seinen substituierten Derivaten, führte zur Entdeckung eines neuen Reaktionswegs, den man als SN2 (aromatisch) bezeichnet. Betrachtet man Chlorbenzol und vergleicht die Reaktionsgeschwindigkeit bei einem nukleophilen Angriff mit der Reaktion von p-Nitrochlorbenzol, so stellt man eine beträchtliche Steigerung der Umsatzgeschwindigkeit fest. Der genaue Mechanismus wird unter dem Artikel Nukleophile aromatische Substitution beschrieben.
SN2t-Mechanismus
Unter einer SN2t-Reaktion versteht man den Angriff eines Nukleophils auf ein sp2-hybridisiertes Kohlenstoffatom, welches besonders stark positiv polarisiert ist. Oft wird diese Reaktion auch als Additions-Eliminierungs-Reaktion an der Carbonsäure oder ihren Derivaten bezeichnet. Es findet dabei eine Umhybridisierung von sp2 zu sp3 statt, weswegen sich ein tetraedrisches Zwischenprodukt bildet (das t in SN2t steht für Tetraedrisch). Anschließend tritt die beste Abgangsgruppe aus und das Kohlenstoffatom rehybridisiert zu sp2. Ein Beispiel hierfür ist die säurekatalysierte Veresterung von Carbonsäuren mit Alkoholen. Die Carboxygruppe wird zunächst protoniert und das Nukleophil, in diesem Fall ein Alkohol, kann angreifen. Nach einer weiteren Protonierung kann Wasser als gute Abgangsgruppe austreten.[7]
SNi-Mechanismus
Die Gewinnung von Alkylchloriden durch nukleophile Substitution von Alkanolen mit Thionylchlorid erfolgt nach einem sogenannten SNi-Mechanismus. Aus einem enantiomerenreinen Alkanol als Edukt wird ein Alkylchlorid mit gleicher Konfiguration erhalten. Die SNi-Reaktion verläuft also unter Retention (Erhalt der Konfiguration). Ob eine SNi-Reaktion oder eine SN2-Reaktion stattfindet, hängt dabei vom Lösungsmittel ab. Das angreifende Nukleophil, in diesem Fall ein Chlorid-Ion, darf nicht im Solvens gelöst sein, deswegen verwendet man bei der SNi-Reaktion Diethylether. Infolge kann das Chlorid-Ion nur intern übertragen werden. Verwendet man dagegen Pyridin als Lösungsmittel, findet eine SN2-Reaktion statt.[8]
Nachbargruppenbeteiligung
Nukleophile Substitutionen können auch durch molekülinterne Prozesse gesteuert werden. So kann es zu einer Beteiligung der schon am betrachteten Kohlenwasserstoff gebundenen Substituenten kommen. Diese intramolekulare Reaktion ist bevorzugt, da die Wahrscheinlichkeit hoch ist, mit den am benachbarten C-Atom liegenden Substituenten zusammenzustoßen. Dieses Nukleophil kann z. B. nicht durch das Lösungsmittel vom Substrat entfernt werden.
Hierbei fungiert die Nachbargruppe (Substituent) als Nukleophil, welches über einen Rückseitenangriff die Abgangsgruppe abspalten lässt. Es bildet sich übergangsweise ein zyklisches System. Ein solcher Zyklus kann einerseits durch eine hohe Ringspannung (kleine Ringe) oder andererseits durch einen Angriff eines externen Nukleophils geöffnet werden. Im zweiten Fall wird demnach unter zweifacher Inversion das Retentionsprodukt erhalten.
Beispiele
Sauerstoff als Nukleophil
- Alkylchloride reagieren mit Hydroxid-Ionen zu Alkoholen unter Freisetzung von Chlorid-Ionen. Analog dazu reagieren chlorierte Aromaten zu Phenolen:
- Alkylchloride reagieren mit Wasser zu protonierten Alkoholen und Chlorid (Hydrolyse):

- Aliphatische Ether und Phenolether können durch nukleophile Substitution von Chlorid durch Alkoholate an Alkyl- oder Arylchloriden gewonnen werden. Diese Reaktion wird auch als Williamson-Ethersynthese bezeichnet.
- Die Synthese von Estern erfolgt durch die Substitution von Chlorid durch Carbonsäuren:

- Arylchloride reagieren mit Cyanat zu Arylcyanaten und Chlorid:

- Aromatische Sulfonsäuren reagieren in Alkalischmelzen zu Phenolen und Sulfit.
Stickstoff als Nukleophil
- Aliphatische primäre Amine entstehen durch den Austausch des Halogenids gegen die Aminogruppe (-NH2). Diese Reaktion findet in Ammoniak als Lösungsmittel statt und wird auch als Ammonolyse bezeichnet.
- Zur Gewinnung sekundärer Amine wird die Reaktion nicht in Ammoniak, sondern mit einem weiteren Amin als Lösungsmittel durchgeführt (→ Aminolyse).
- Tertiäre Amine entstehen durch die Umsetzung mit einem sekundären Amin,
- Tetraalkylammoniumsalze durch die Umsetzung mit einem tertiären Amin.
- Zur Gabriel-Synthese gehört eine Reaktion, bei der ein Alkylchlorid oder -bromid mit dem Phthalimidanion umgesetzt wird:

Schwefel als Nukleophil
- Die Reaktionen von Alkyl- und Arylhalogeniden mit Hydrogensulfid und Thiolaten führen analog zu denen mit den Sauerstoff-Homologen Hydroxid und Alkoholaten zu Thiolen und Thioethern.
- Mit Thioharnstoff reagieren Alkylhalogenide zu Isothiuronium-Salzen.
- Durch Substitution des Halogens mit Hydrogensulfit entstehen Sulfonsäuren.
Halogenide als Nukleophil
- Werden Alkyl- oder Arylchloride beziehungsweise -bromide mit einem Überschuss an Fluorid (in polaren, aprotischen Lösungsmitteln) oder Iodid (in Aceton) umgesetzt, entstehen aliphatische oder aromatische Fluoride oder Iodide. Die Reaktion mit Iodid wird als Finkelstein-Reaktion bezeichnet.
Phosphor als Nukleophil
- Alkylchloride reagieren mit Alkyl- oder Arylphosphanen zum entsprechenden Phosphoniumsalz. Aus organischen Phosphoniumsalzen werden die Olefinierungsreagenzien für die Wittig-Reaktion gewonnen.

Hydrid als Nukleophil
- Alkane können durch Reaktion von Alkylhalogeniden mit Hydrid als Substituent hergestellt werden. Hydrid-Donator ist Lithiumaluminiumhydrid.
 Reaktion von Alkylchloriden mit Lithiumaluminiumhydrid
Reaktion von Alkylchloriden mit Lithiumaluminiumhydrid
Die Eliminierungsreaktion als mögliche Konkurrenzreaktion
E1 steht in Konkurrenz zur SN1-Reaktion, E2 zur SN2-Reaktion. Steuern kann man dies unter anderem über Lösungsmitteleinflüsse.
siehe auch: Eliminierungsreaktion
Literatur
- Organikum, 16. Auflage, VEB Deutscher Verlag der Wissenschaften Berlin 1985, ISBN 3-326-00076-6.
- Andrew Streitwieser Jr., Clayton H. Heathcock, Organische Chemie, VCH Weinheim 1980, ISBN 3-527-25810-8.
- Peter Sykes: Reaktionsmechanismen – eine Einführung, 8. Auflage VCH Weinheim 1982 ISBN 3-527-21090-3.
Einzelnachweise
- Siegfried Hauptmann: Reaktion und Mechanismus in der organischen Chemie, B. G. Teubner, Stuttgart, 1991, S. 78, ISBN 3-519-03515-4.
- Ivan Ernest: Bindung, Struktur und Reaktionsmechanismen in der organischen Chemie, Springer-Verlag, 1972, S. 107–111, ISBN 3-211-81060-9.
- Paula, Yurkanis, Bruice: Organic Chemistry. 4. Auflage, Prentice-Hall, 2003, ISBN 0-13-141010-5, S. 403–447.
- K. P. C. Vollhardt, Neil E. Schore: Organische Chemie, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005, 4. Auflage, H. Butenschön, S. 248–296, ISBN 3-527-31380-X.
- Doi, Togano, Xantheas, Nakanishi, Nagata, Ebata, Inokuchi: Microhydration Effects on the Intermediates of the SN2 Reaction of Iodide Anion with Methyl Iodide. In: Angewandte Chemie, 125, 16, 1521–3757, doi:10.1002/ange.201207697.
- Ulrich Lüning: Organische Reaktionen – Eine Einführung in der Reaktionswege und Mechanismen. 2. Auflage. Spektrum, München 2007, ISBN 978-3-8274-1834-0, S. 45.
- Helmut Wachter: Chemie für Mediziner, Walter de Gruyter, 8. Auflage, S. 323, ISBN 978-3-11-017581-3.
- Reinhard Brückner: Reaktionsmechanismen, Spektrum Akademischer Verlag, 3. Auflage, S. 93, ISBN 978-3-8274-1579-0.
Weblinks
- Animation: Der SN1-Mechanismus
- Animation: Der SN2-Mechanismus