Säurekonstante
Die Säurekonstante KS ist eine Stoffkonstante, die Aufschluss darüber gibt, in welchem Maße ein Stoff in einer Gleichgewichtsreaktion mit Wasser unter Protolyse reagiert:[1]
- .

.png.webp)
Dabei steht HA für eine Brønsted-Säure (nach Johannes Nicolaus Brønsted), die ein Proton H+ an ein Lösungsmittel wie Wasser abgeben kann, wobei ein Anion A− zurückbleibt.[2] Allgemeiner gilt die Brønsted'sche Definition auch für nichtwässrige Systeme, hier gilt für ein beliebiges, protonierbares Lösungsmittel Y:
- .

KS ist die mit [Y] multiplizierte Gleichgewichtskonstante dieser Reaktion und damit ein Maß für die Stärke einer Säure.[1] Je stärker die Säure, desto mehr ist die Reaktion auf die rechte Seite verschoben; d. h., umso höher sind die Konzentrationen von HY+ und A−. Die Gleichgewichtskonstante wird meist als ihr negativer dekadischer Logarithmus, der pKS-Wert, angegeben (auch pKa, von engl. acid = Säure). Das bedeutet: Je kleiner der pKS-Wert ist, desto stärker ist die Säure.
Herleitung der Säurekonstanten
Die Säurekonstante leitet sich als Gleichgewichtskonstante einer chemischen Reaktion aus der Gibbs-Energie (auch Freie Enthalpie) her. Ist diese bekannt, so gilt für die Gleichgewichtskonstante einer beliebigen chemischen Reaktion:


- ,

wobei die Universelle Gaskonstante, die Temperatur und die eulersche Zahl ist. Die Formel beschreibt so auch die beobachtbare Temperaturabhängigkeit der Säurekonstanten.



ist dabei als Produkt der Aktivitäten definiert und ist eine dimensionslose Größe. Werden Mischungseffekte vernachlässigt, gilt . Dies ist in Lösungen bis 1 mmol/l ohne größere Fehler möglich. Konstanten können daher mit den Aktivitäten wie auch mit den Konzentrationen aufgestellt werden. Sie besitzen jedoch einen anderen Zahlenwert. Bedingt durch die historische Entwicklung der Chemie aus einer praktischen Wissenschaft werden meist die konzentrationsbezogenen Konstanten angegeben, da diese experimentell vor der thermodynamischen Begründung gefunden wurden.

Säurestärke
Die Eigenschaft eines bestimmten Stoffes, als Säure zu reagieren, ist untrennbar verknüpft mit seiner potentiellen Fähigkeit, ein Proton (H+) an einen Reaktionspartner zu übertragen. Man nennt eine solche Reaktion Protolyse. Die Stärke einer Säure beschreibt das Ausmaß dieser Fähigkeit. Diese ist jedoch abhängig von der Fähigkeit eines Reaktionspartners, das Proton aufzunehmen. Soll die Säurenstärke verschiedener Säuren verglichen werden, ist es sinnvoll, die Wechselwirkung mit einem Standardreaktionspartner zu betrachten. Dieser ist in der Regel das Wasser, das auch in vielen Vorgängen in der Natur die bedeutsamste Verbindung und Lösemittel ist. Die Reaktionsgleichung einer Säure HA in und mit Wasser kann so dargestellt werden:

In dieser Reaktion stellt sich schnell ein Gleichgewicht ein. Hier verfügt neben HA auch H3O+ über die Fähigkeit, ein Proton an einen Reaktionspartner zu übertragen: Sie sind beide Säuren. H2O und auch A− haben hingegen die Fähigkeit, ein Proton aufzunehmen, weswegen man sie beide als Basen bezeichnet. Trennt man gedanklich die Standardreaktionspartner Wasser und H3O+ ab, bleiben HA und A− übrig. Da die Konzentrationen dieser Komponenten an ein Gleichgewicht gebunden sind, ist das Ausmaß der Fähigkeit von HA, eine Säure zu sein, gekoppelt an das Ausmaß der Fähigkeit von A−, eine Base zu sein. Hat beispielsweise HA ein großes Potenzial, ein Proton abzugeben und A− ein kleines Potenzial, ein Proton anzunehmen, nennt man HA eine starke Säure. Das Gleichgewicht (1) würde auf der rechten Seite stehen. Wenn die Säure HA ein großes Potential hat, ein Proton abzugeben (also einen niedrigen pKS-Wert), dann hat dessen korrespondierende Base A− ein in dem Maße geringes Potential (also einen hohen pKB-Wert), ein Proton aufzunehmen. Für Wasser z. B. gilt: pKB + pKS = pKW = 14
Die Säurekonstante (bzw. der pKS-Wert) ist ein Maß für die Stärke einer Säure. Die Acidität ist umso größer, je geringer ihr pKS-Wert ist. Der pKS-Wert ist numerisch gleich dem pH-Wert einer Lösung, wenn HA und A− nach Gleichgewicht (1) in gleicher Konzentration vorliegen.
In wässrigen Lösungen protolysieren sehr starke Säuren und sehr starke Basen vollständig zu H3O+- bzw. OH−-Ionen. So lassen sich die unterschiedlichen Säurestärken von Chlorwasserstoff und Perchlorsäure in Wasser nicht mehr anhand des pH-Wertes unterscheiden. Hier spricht man vom nivellierenden Effekt (v. frz.: niveler = gleichmachen) des Wassers. Um auch sehr starke Säuren bezüglich der Säurestärke unterscheiden zu können, bestimmt man Gleichgewichtskonstanten in nichtwässrigen Lösungen und überträgt diese annäherungsweise auf das Lösungsmittel Wasser.
Der Standardreaktionspartner Wasser hat die besondere Eigenschaft, als Säure und Base reagieren zu können:

Diese sogenannte Autoprotolyse erlaubt die Bestimmung des Ausmaßes der Fähigkeit einer Base, ein Proton vom Wasser zu übernehmen, und wird unter Basenkonstante näher erläutert.
Ursachen der verschiedenen Säurestärken
Die Säurestärke eines Moleküls kann an verschiedenen Faktoren abgeschätzt werden.
Eine Säure ist umso stärker bzw. gibt umso leichter ein Proton ab,
- wenn ein induktiver Elektronenzug vorhanden ist (−I-Effekt).
- je stabiler die korrespondierende Base ist, das heißt je schwächer die korrespondierende Base ist.
- wenn das elektronegativere Atom das dissoziierbare Wasserstoffatom trägt (bei Atomen gleicher Größe).
- wenn das größere Atom das Wasserstoffatom trägt (bei Atomen verschiedener Größe).
- je niedriger die Standardbildungsenthalpie ist.
- je instabiler das Säure-Molekül ist.[3]
Säure-Base-Reaktion
Zwischen einer Säure HA und ihrer Base A− liegt in wässriger Lösung folgende Gleichgewichtsreaktion vor:
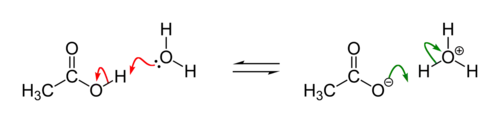
Rote Pfeile: Deprotonierung der Essigsäure
Grüne Pfeile: Protonierung des Acetats unter Bildung von Essigsäure.
Nach dem Massenwirkungsgesetz wird die Lage des Gleichgewichtes durch die Gleichgewichtskonstante K beschrieben:

Da die Konzentration von Wasser (c(H2O)) bei der Reaktion praktisch konstant bleibt, lässt sich c(H2O) in die Konstante K einbeziehen. Damit ergibt sich schließlich die Säurekonstante KS mit der Einheit mol/l:

Häufig wird der negative dekadische Logarithmus von KS, der so genannte pKS-Wert, angegeben:

Je kleiner der pKS-Wert, desto stärker ist die Säure. So hat zum Beispiel Salpetersäure (HNO3, Dissoziationsgrad von 96 % bei einer Konzentration von 1 mol/l) den pKS-Wert −1,32, Essigsäure (Dissoziationsgrad von 0,4 % bei einer Konzentration von 1 mol/l) einen pKS-Wert von 4,75.
Entsprechend gibt es eine Basenkonstante (pKB-Wert). Je kleiner der pKB-Wert, desto stärker das Bestreben der Base, Protonen aufzunehmen. Den pKS-Wert kann man auf die Basenkonstante der korrespondierenden Base umrechnen:
- .

Säure- und Basenkonstanten sind temperaturabhängig. In der Regel werden die Konstanten bei Temperaturen im Bereich von 23 bis 25 Grad Celsius bestimmt. In diesem Bereich ergibt das Ionenprodukt des Wassers ausreichend genau
- .

Mehrprotonige Säuren
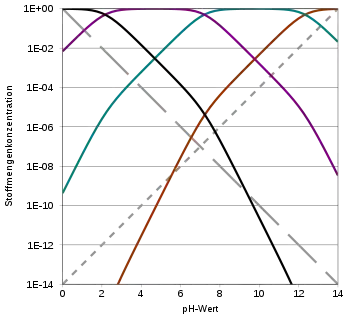
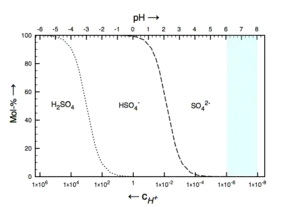
| H2SO4 | HSO4− | SO42− |
| H+ | OH− |
Bei einer mehrprotonigen Säure besteht eine schrittweise Protolyse. Für jede Protolysationsstufe liegt eine eigene Säurekonstante bzw. pKS-Wert vor. Für die einzelnen Protolyseschritte gilt im Allgemeinen: KS1 > KS2 > KS3 (bzw. pKS1 < pKS2 < pKS3), da aus der steigenden Ionenladung des entstehenden Säurerestanions die weiterführende Protolyse weniger energetisch begünstigt ist.
Als Beispiel gilt für die Phosphorsäure:









Beim pH-Wert 7,20 liegen näherungsweise gleich große Konzentrationen an Dihydrogen- und Hydrogenphosphat-Ionen vor, die Konzentrationen an undissoziierter Phosphorsäure und Phosphationen sind millionenfach kleiner. Diese Zusammenhänge macht man sich in Phosphatpuffern zu Nutze.
Schwefelsäure ist um fünf Größenordnungen acider als Phosphorsäure:






Konzentrierte Schwefelsäure wird in Blei-Akkumulatoren als Elektrolyt verwendet. Unter diesen Gleichgewichtsbedingungen existieren keine freien Sulfat-Ionen mehr.
Bestimmung von pKS-Werten
Die pKS-Werte von Säuren mit Werten im Bereich von 4 bis etwa 10 lassen sich über Säure-Base-Titrationen und die Bestimmung des pH-Werts am Halbäquivalenzpunkt bestimmen. Hier liegen die Säure und ihre korrespondierende Base in gleicher Konzentration vor. An diesem Punkt folgt aus der Henderson-Hasselbalch-Gleichung: pH = pKS.
Acidität organischer Säuren
Bei organischen Säuren entscheiden vor allem drei Struktureigenschaften über die Säurestärke:
- Stabilisierung des entstehenden Anions durch Mesomerie: So sind z. B. Carbonsäuren saurer als Alkohole. Mesomere Effekte spielen hierbei eine entscheidende Rolle: Ein −M-Effekt (etwa einer Nitrogruppe –NO2) erhöht die Säurestärke, ein +M-Effekt verringert sie.[4]
- Hybridisierung des Kohlenstoffatoms: Mit steigendem s-Gehalt nimmt die Stärke zu. So hat Ethin (sp-Hybridorbital) einen niedrigeren pKS-Wert als Ethen (sp2-Hybridorbital) und dieses einen niedrigeren als Ethan (sp3-Hybridorbital), es gilt also für den pKS-Wert: sp < sp2 < sp3; die Werte betragen für Ethin 25, für Ethen 44 und für Ethan 50.[5]
- Induktive Effekte: Die Säurestärke steigt, wenn elektronenziehende Gruppen vorhanden sind, z. B. Halogene wie Fluor und Chlor oder Sauerstoff. Trichloressigsäure ist beispielsweise eine stärkere Säure als Essigsäure.
Einige Substituenten besitzen sowohl mesomere als auch induktive Effekte, etwa die Halogene oder Nitrogruppen. Halogene weisen einen starken −I-, aber einen schwachen +M-Effekt auf; die Nitrogruppe wirkt sowohl elektronenanziehend (−I-Effekt) als auch über einen −M-Effekt, d. h. beide Effekte wirken in dieselbe Richtung.[4]
pKS- und pKB-Werte einiger Verbindungen
Die folgende Tabelle listet pKS- und pKB-Werte einiger Säuren und Basen bei Standardbedingungen:[6]
| Säurestärke | pKS | Säure + H2O H3O+ + Base | pKB | Basenstärke | |
|---|---|---|---|---|---|
| sehr stark | −17 | H[SbF6] | [SbF6]− | 31 | |
| −10 | HClO4 | ClO4− | 24 | sehr schwach | |
| −10 | HI | I− | 24 | ||
| −8,9[7] | HBr | Br− | 22,9 | ||
| −6 | HCl | Cl− | 20 | ||
| −3 | H2SO4 | HSO4− | 17 | ||
| −1,32 | HNO3 | NO3− | 15,32 | ||
| stark | 0,00[8][9] | H3O+ | H2O | 14,00 | schwach |
| 1,92 | HSO4− | SO42− | 12,08 | ||
| 2,13 | H3PO4 | H2PO4− | 11,87 | ||
| 2,22 | [Fe(H2O)6]3+ | [Fe(OH)(H2O)5]2+ | 11,78 | ||
| 3,14 | HF | F− | 10,86 | ||
| 3,75 | HCOOH | HCOO− | 10,25 | ||
| mittelstark | 4,75 | CH3COOH | CH3COO− | 9,25 | mittelstark |
| 4,85 | [Al(H2O)6]3+ | [Al(OH)(H2O)5]2+ | 9,15 | ||
| 6,52 | H2CO3 | HCO3− | 7,48 | ||
| 6,92 | H2S | HS− | 7,08 | ||
| 7,20 | H2PO4− | HPO42− | 6,80 | ||
| schwach | 9,25 | NH4+ | NH3 | 4,75 | stark |
| 9,40 | HCN | CN− | 4,60 | ||
| 9,8 | Trimethyl-Ammonium | Trimethylamin | 4,2 | ||
| 10,40 | HCO3− | CO32− | 3,60 | ||
| 10,6 | Methyl-Ammonium | Methylamin | 3,4 | ||
| 10,73 | Dimethyl-Ammonium | Dimethylamin | 3,27 | ||
| 12,36 | HPO42− | PO43− | 1,64 | ||
| 13,00 | HS− | S2− | 1,00 | ||
| 14,00 | H2O | OH− | 0,00 | ||
| sehr schwach | 15,90 | CH3-CH2-OH | CH3-CH2-O− | −1,90 | sehr stark |
| 23 | NH3 | NH2− | −9 | ||
| 48[10] | CH4 | CH3− | −34 | ||

Weblinks
- Bordwell pKS Tabelle in DMSO
- pKS Data (PDF; 645 kB), Compiled by R. Williams, auf chem.wisc.edu.
- Harvard University: Evans Group pKS Tabelle (PDF; 240 kB).
- Eintrag zu Acidity constant. In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.A00080 – Version: 2.3.1.
- pKS Tabelle CCI ETH
- pKS Tabelle mit sehr vielen Substanzen
- Umfangreiche Tabellen mit Säuren und Basen (MS Excel; 1,2 MB)
- Interaktiv sortierbare, sehr umfangreiche Liste mit pKS-Werten organischer und anorganischer Säuren und Basen
{kind=link}
Einzelnachweise
- Eintrag zu acidity constant. In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.A00080 – Version: 2.3.1.
- Wissenschaft-Online-Lexika: Eintrag zu Säure-Base-Konzepte im Lexikon der Chemie, abgerufen am 2. April 2008.
- Bruice, P.Y.: Organische Chemie: Studieren kompakt. 5., aktualisierte Auflage. Pearson Studium, München 2011, ISBN 978-3-86894-102-9, S. 53–60.
- Alfons Hädener, Heinz Kaufmann: Grundlagen der organischen Chemie. 11. überarbeitete und erweiterte Auflage. Birkhäuser, Basel u. a. 2006, ISBN 3-7643-7040-8.
- Michael B. Smith and Jerry March, March's Advanced Organic Chemistry, John Wiley and Sons, 2007, ISBN 0-471-720-91-7
- Gerhart Jander, Karl Friedrich Jahr, Gerhard Schulze, Jürgen Simon (Hrsg.): Maßanalyse. Theorie und Praxis der Titrationen mit chemischen und physikalischen Indikationen. 16. Auflage. Walter de Gruyter, Berlin u. a. 2003, ISBN 3-11-017098-1, S. 81.
- Eberhard Gerdes: Qualitative Anorganische Analyse: Ein Begleiter für Theorie und Praxis. Springer DE, 2000, ISBN 3-540-67875-1, S. 154 (eingeschränkte Vorschau in der Google-Buchsuche).
- P.W. Atkins, T.L. Overton, J.P. Rourke, M.T. Weller, F.A. Armstrong: Shriver & Atkins´ inorganic chemistry. 5th Edition. Oxford University Press, Oxford New York 2010, ISBN 978-0-19-9236176, S. 115.
- A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 91.–100., verbesserte und stark erweiterte Auflage. Walter de Gruyter, Berlin 1985, ISBN 3-11-007511-3, S. 241.
- Jerry March: Advanced Organic Chemistry. Reactions, Mechanisms, and Structure. 3. Auflage. Wiley, New York NY u. a. 1985, ISBN 0-471-88841-9, S. 222.