Aktivierungsenergie
Die Aktivierungsenergie, geprägt 1889 von Svante Arrhenius, ist eine energetische Barriere, die bei einer chemischen Reaktion von den Reaktionspartnern überwunden werden muss. Allgemein gilt: Je niedriger die Aktivierungsenergie, desto schneller verläuft die Reaktion. Eine hohe Aktivierungsenergie hemmt Reaktionen, die wegen fester Bindung der Endprodukte aus energetischen Gründen zu erwarten wären, und verhindert oder verzögert damit die Einstellung eines chemischen Gleichgewichts. So kann eine Mischung aus Methan und dem Sauerstoff der Luft bei Standardbedingungen nahezu unverändert existieren (d. h. die Reaktion läuft unmessbar langsam ab), obwohl die exergone Reaktion zu Kohlendioxid und Wasser aus thermodynamischer Sicht „spontan“ ablaufen sollte. Die Aktivierungsenergie nach Arrhenius ist eine empirische Größe, die sich durch die hohe Temperaturabhängigkeit der Geschwindigkeit von vielen chemischen Reaktionen ermitteln lässt.[1]
Allgemein werden alle Prozesse als aktivierte Prozesse bezeichnet, bei denen eine energetische Barriere überwunden werden muss, um ein thermodynamisches System von einem Zustand in einen anderen zu überführen. Hierzu zählen neben chemischen Reaktionen im engeren Sinne auch zahlreiche andere Prozesse wie Änderungen der Konformation, Keimbildung, Kristallisation, Kavitation oder die Entstehung von Brüchen oder Erdbeben.[2]
Reaktionskinetik
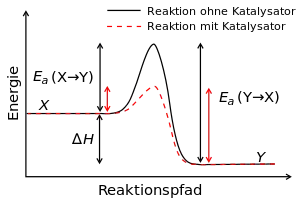
In der Vorstellung der physikalischen Chemie muss im Verlauf einer chemischen Reaktion eine Umgruppierung der Atome aus der Anordnung der Reaktanten in die der Produkte stattfinden, wobei alte Bindungen aufgebrochen und neue Bindungen geknüpft werden. Die Reaktanten durchlaufen dabei für die Umwandlung in deren Produkte einen aktivierten Zustand, den sogenannten Übergangszustand (siehe Abbildung rechts, Kurvenmaximum), dessen Bildung eine bestimmte Energie (Aktivierungsenergie) erfordert. Die Geschwindigkeit einer chemischen Reaktion hängt also (bei konstanter Temperatur), nicht von den Energien der Reaktanten und Produkte, sondern nur von der Energiedifferenz zwischen Reaktanten und dem Übergangszustand ab. Je höher die Temperatur des Reaktionssystems, umso höher ist auch die Wahrscheinlichkeit, dass die Reaktanten die benötigte Aktivierungsenergie bereitstellen, die Energiebarriere überwinden und zum Produkt weiterreagieren.
Temperaturabhängigkeit der Reaktionsgeschwindigkeit
Der Zusammenhang zwischen der Geschwindigkeitskonstanten , der Aktivierungsenergie und der thermodynamischen Temperatur kann (unter Vernachlässigung des Aktivierungsvolumens bzw. bei niedrigen Drücken) in vielen Fällen durch die Arrhenius-Gleichung[3][4] mit dem präexponentiellen Frequenzfaktor und der Gaskonstante beschrieben werden:






Logarithmiert man die Gleichung, so ergibt sich:

Da der präexponentielle Faktor häufig genügend unabhängig von der Temperatur ist, gilt für den Zahlenwert von :


Gelingt die Bestimmung der Geschwindigkeitskonstanten von irreversiblen Reaktionen bei unterschiedlichen Temperaturen, kann gegen aufgetragen werden und aus der Steigung der Geraden bestimmt werden; siehe dazu Arrheniusgraph.


Sind zwei Geschwindigkeitskonstanten ( und ) einer Reaktion bei zwei Temperaturen ( und ) bekannt, lässt sich mit





die Aktivierungsenergie berechnen.[5] Die Gleichung ist eine umgeformte Differenz aus zwei logarithmierten Arrhenius-Gleichungen (je eine pro Temperatur). Bei vielen Reaktionen in Lösung liegt die Aktivierungsenergie im Bereich von 50 kJ·mol−1.[6] Eine Temperaturerhöhung von 290 K auf 300 K führt dabei annähernd zu einer Verdoppelung der Geschwindigkeitskonstanten (siehe RGT-Regel). Es ist jedoch stets zu beachten, dass mit zunehmender Aktivierungsenergie (d. h. mit zunehmender Steigung im Arrheniusgraph) der Effekt der Temperaturabhängigkeit verstärkt wird. So werden Reaktionen mit niedrigen Aktivierungsenergien (ca. 10 kJ·mol−1) durch Temperaturerhöhung nur geringfügig beschleunigt. Die Geschwindigkeit von Reaktionen mit großen Aktivierungsenergien (ca. 60 kJ·mol−1) nimmt hingegen mit steigender Temperatur stark zu. Die Werte für die molaren Aktivierungsenergien vieler gängiger Reaktionen liegen zwischen 30 und 100 kJ·mol−1.[7]
Bei einigen Reaktionen folgt die Temperaturabhängigkeit der Geschwindigkeitskonstante nicht der Arrhenius-Gleichung. Dies sind beispielsweise Reaktionen ohne Aktivierungsenergie, explosionsartige Reaktionen, Reaktionen mit vorgelagerten Gleichgewichten und viele enzymatische oder heterokatalytische Reaktionen.[8]
Theoretischer Hintergrund
Tatsächlich beschreibt das Modell von Arrhenius die Vorgänge bei einer chemischen Reaktion unvollständig. Die Arrhenius-Gleichung lässt sich durch die klassische Stoßtheorie theoretisch begründen. Die hohe Wirkung einer Temperaturerhöhung auf die Geschwindigkeit einer Reaktion beruht auf der starken Zunahme des Anteils der Teilchen, die über genug Energie verfügen, um die Barriere zu überwinden. Nebenbei wächst bei einer Temperaturerhöhung auch die Häufigkeit der Zusammenstöße (die Stoßzahl) der Reaktanden. Der Anstieg der Stoßzahl führt praktisch jedoch zu einer sehr geringen Erhöhung der Reaktionsgeschwindigkeit und geht in der Arrhenius-Gleichung als Komponente im „temperaturunabhängigen“ präexponentiellen Faktor A unter. Ist die Aktivierungsenergie klein oder null bestimmen die Stoßzahl oder die Diffusionsgeschwindigkeit die Reaktionsgeschwindigkeit. Nach Eyring (siehe auch Theorie des Übergangszustandes) ist die Freie Aktivierungsenthalpie die bestimmende Größe für die Reaktionsgeschwindigkeit.

Katalyse
Ein Katalysator setzt die Aktivierungsenergie für chemische Reaktionen herab, ändert jedoch nicht die freie Reaktionsenthalpie . Man nimmt an, dass bei Anwesenheit eines Katalysators ein Komplex mit niedrigerer Aktivierungsenergie gebildet wird und so die Reaktionswahrscheinlichkeit steigt.

Siehe auch
Literatur
- Siehe Kinetik (Chemie)
Einzelnachweise
- Eintrag zu activation energy (Arrhenius activation energy). In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.A00102 – Version: 2.3.2.
- Jan Wedekind, Reinhard Strey: New method to analyze simulations of activated processes. In: Journal of Chemical Physics, 2007, Band 126, 134103, doi:10.1063/1.2713401.
- Jacobus Henricus van't Hoff, Études de dynamique chimique, Frederik Muller & Co., Amsterdam 1884, S. 114–118.
- Svante Arrhenius, Z. Phys. Chem. 1889, 4, S. 226–248.
- Hans Rudolf Christen: Grundlagen der allgemeinen und anorganischen Chemie, Otto Salle, Frankfurt a. M., Sauerländer, Aarau, 9. Auflage, 1988, S. 333.
- Hans Kuhn, Horst-Dieter Försterling: Principles of Physical Chemistry, John Wiley, Chichester, 1999, S. 683.
- Georg Job, Regina Rüffler: Physikalische Chemie, Vieweg+Teubner Verlag, Wiesbaden, 1. Auflage 2011, S. 401–402.
- Gerd Wedler: Lehrbuch der Physikalischen Chemie, VCH, Weinheim, 3. Auflage, 1987, S. 169.
Weblinks
- Verzeichnis von Nachschlagewerken und Datenbanken mit Aktivierungsenergien
- Video: Wie bestimmt man die Aktivierungsenergie einer Reaktion? - Auswertung nach ARRHENIUS. Jakob Günter Lauth (SciFox) 2013, zur Verfügung gestellt von der Technischen Informationsbibliothek (TIB), doi:10.5446/15614.