Pericyclische Reaktion
Pericyclische Reaktionen sind chemische Reaktionen, bei denen die Bindungsverhältnisse durch eine konzertierte Verschiebung von Elektronen verändert werden, ohne dass radikalische oder ionische Zwischenstufen auftreten. Die dabei durchlaufenen Übergangszustände sind cyclischer Natur.
Die wichtigsten pericyclischen Reaktionen sind
- elektrocyclische Reaktionen
- Cycloadditionen
- sigmatrope Umlagerungen
- Cheletrope Reaktionen
Systematik
Es existieren insgesamt vier Konzepte zur Klärung der Reaktivität von pericyclischen Reaktionen:
- Orbitalkorrelationsdiagramme nach Robert Burns Woodward und Roald Hoffmann[1]
- Methode des obersten besetzten Orbitals nach Robert Burns Woodward und Roald Hoffmann
- Grenzorbitalmethode nach Kenichi Fukui[2]
- Konzept der aromatischen Übergangszustände nach M. J. S. Dewar, H. Zimmerman und H. Evans
Die Grenzorbitalmethode eignet sich nur zur Klärung bi- oder höhermolekularer Reaktionen, Orbitalkorrelationsdiagramme und die Methode des obersten besetzten Orbitals nur für monomolekulare oder intramolekulare Reaktionen. Das Konzept des aromatischen Übergangszustandes ist allgemein gültig. Hierbei erfolgt die Stabilisierung im cyclischen Übergangszustand in der Regel über 3 Elektronenpaare. Sechselektronen-Reaktionen lassen sich über ein Sechszentren- oder Fünfzentren-Modell (mit einem nichtbindenden Elektronenpaar) beschreiben, wobei die σ-Bindungen als reagierendes Spezies fungieren. Daneben gibt es noch weitere pericyclische Systeme, wie beispielsweise Dreizentren-Zweielektronen-Systeme oder Zehnelektronen-Systeme.[3]
Reaktionen
Elektrocyclische Reaktionen
Bei elektrocyclischen Reaktionen kommt es zu Ringschlüssen zwischen den Enden eines linearen konjugierten -Systems, zum Beispiel 1,3-Butadien. Auch längerkettige oder substituierte konjugierte Systeme reagieren in dieser Art. Es gibt zwei Möglichkeiten des Ringschlusses: Entweder konrotatorisch oder disrotatorisch. Im ersten Fall drehen sich die Substituenten an den terminalen C-Atomen in die gleiche Richtung während der Bildung der neuen Bindung, im letzten Fall in entgegengesetzte Richtung. Eine Erklärungsmöglichkeit sind Orbitalkorrelationsdiagramme. Man stellt hierfür alle bindenden und antibindenden Molekülorbitale von Edukt und Produkt in energetischer Reihenfolge dar. Dann korreliert man Orbitale gleicher Symmetrie von Edukt und Produkt miteinander. Ist es möglich nur bindende und nur antibindende Orbitale zu korrelieren, ist der Vorgang thermisch erlaubt und die Reaktion kann stattfinden. Da die Reaktion konzertiert ablaufen soll, können dann aus den Eduktmolekülorbitalen kontinuierlich Produktmolekülorbitale entstehen. Sollte eine Korrelation von einem oder mehreren bindenden mit antibindenden Orbitalen vorkommen, ist die Reaktion thermisch verboten und findet nicht statt. Jedoch ist die Reaktion dann photochemisch möglich, denn ein angeregter Zustand besitzt eine andere Orbitalsymmetrie. Für den disrotatorischen Prozess haben die Produktmolekülorbitale eine andere Symmetrie als bei einem konrotatorischen Prozess. Es zeigt sich, dass beispielsweise die Cyclisierung von Butadien zu Cyclobuten unter thermischen Bedingungen nur konrotatorisch möglich ist.

Zum gleichen Ergebnis kommt man mit der Methode des obersten besetzten Orbitals. Man betrachtet die Vorzeichen bzw. die Symmetrie des HOMO des Edukts. Man sucht die Drehung der Orbitallappen, die zu einer konstruktiven Überlappung führen würden, also man dreht so, dass sich Lappen mit gleichem Vorzeichen treffen. Auch hier gilt, dass man photochemisch zum entgegengesetzten Ergebnis kommt, weil der angeregte Zustand eine Knotenebene mehr im HOMO hat und damit genau die entgegengesetzte Symmetrie.
Bei der Cycloheptatrien-Norcaradien-Umlagerung ist die Lage des Gleichgewichtes von der Natur des Atoms bzw. der Gruppe X abhängig:[4]
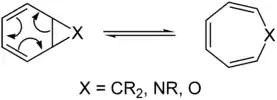
Cycloadditionen
Bei Cycloadditionen kommt es zu einem Ringschluss durch die Verknüpfung der Enden zweier -Systeme. Man unterscheidet grundsätzlich zwischen einer suprafacialen Annäherung der Edukte aneinander und einer antarafacialen Annäherung. Im ersten Fall findet die Addition durch den Reaktanten auf derselben Seite des Substrates statt, im letzten Fall wird eine Bindung auf der einen und eine auf der anderen Seite des Substrates gebildet. Derartige Antara-Additionen sind sterisch sehr ungünstig, sodass im Allgemeinen nur Supra-Additionen stattfinden. Ein klassisches Beispiel für eine suprafaciale Cycloaddition ist die Diels-Alder-Reaktion.
Solche Reaktionen können mit der Grenzorbitalmethode erklärt werden. Man betrachtet hierbei das LUMO des einen Reaktanten und das HOMO des anderen. Um die Stereochemie und die Möglichkeit der Reaktion voraussagen zu können, nähert man die Reaktanten so an, dass es zu einer konstruktiven Überlappung der entsprechenden Grenzorbitale kommt; d. h. Orbitallappen mit gleichem Vorzeichen müssen überlappen. Ist dies möglich, so findet die Reaktion unter thermischen Bedingungen statt, ansonsten ist Photochemie vonnöten.
Cheletrope Reaktionen
Sie sind ein Sonderfall der Cycloadditionen, bei denen die neu geknüpften Bindungen vom selben Atom ausgehen. Beispielhaft ist dies die Addition von Carbenen an Doppelbindungen. Auch diese Reaktionen sind über die Grenzorbitalmethode nachvollziehbar.
Sigmatrope Umlagerungen
Sigmatrope Umlagerungen sind Reaktionen, bei denen es intramolekular zur Lösung einer -Bindung kommt, die an einer anderen Stelle wiederhergestellt wird. Ein Beispiel für eine sigmatrope Umlagerung ist die Cope-Umlagerung.

Diese Reaktionen werden mit der Methode des obersten besetzten Orbitals erklärt, da es hier nur zu Wechselwirkungen zwischen den HOMOs der wandernden Gruppe und des Restgerüstes kommt. Die Stereochemie ist somit von der Symmetrie der beteiligten Orbitale abhängig. Abhängig von der Zahl der Knoten im Molekülorbital des theoretisch intermediär auftretenden Polyenylradikals, findet die Wasserstoffverschiebung suprafacial oder antarafacial statt. Je nachdem, mit welchen Orbitalhälften das s-Orbital des Wasserstoffs konstruktiv überlappen kann. Sollte eine Kohlenstoffgruppe wandern, so sind theoretisch beide Wege möglich, da das p-Orbital am Kohlenstoff zwei Lappen mit entgegengesetztem Vorzeichen besitzt. Somit können Kohlenstoffverschiebungen unter Retention der Konfiguration verlaufen, wenn nur eine Orbitalhälfte der wandernden Gruppe beteiligt ist oder unter Konfigurationsinversion, wenn beide Orbitalhälften beteiligt sind. Im Allgemeinen ist die antarafaciale Kohlenstoffverschiebung aber geometrisch sehr ungünstig, sodass es bei unterschiedlichen Vorzeichen am Ende des Polyenylradikals zur antarafacialen Umlagerung unter Konfigurationsinversion und bei gleichem Vorzeichen zur suprafacialen Umlagerung unter Konfigurationsretention kommt. Kohlenstoffverschiebungen sind jedoch aufgrund der hohen Masse der wandernden Gruppe sehr selten und die Wasserstoffverschiebung wird bevorzugt.
Konzept des aromatischen Übergangszustandes
Wenn der Übergangszustand einer pericyclischen Reaktion der Topologie eines (Hückel-)Aromaten sehr nahekommt, und die Anzahl der beteiligten Elektronen denen eines Aromaten entspricht, so ist die Reaktion thermisch erlaubt. Weiterhin führen auch möbiusaromatische Übergangszustände zu thermisch erlaubten Reaktionen. In letzteren kommt es zu einer ungeraden Anzahl von Phasenumkehrungen im obersten besetzten Orbital. Hückelaromatische Übergangszustände besitzen dagegen keine oder eine gerade Anzahl von Phasenumkehrungen. Zusammenfassend lässt sich feststellen: Wenn die Summe aus der Anzahl der an der Reaktion beteiligten Bindungen und der Anzahl der Phasenumkehrungen im Übergangszustand eine ungerade Zahl ergibt, so ist die Reaktion thermisch erlaubt. Die Phasenumkehrungen bestimmen die Topologie des Übergangszustandes und damit die Stereochemie der Reaktion.
Siehe auch: Macrophomat-Synthase
Einzelnachweise
- R. B. Woodward, Roald Hoffmann: The Conservation of Orbital Symmetry. In: Angewandte Chemie International Edition. Band 8, Nr. 11, 1969, S. 781–853, doi:10.1002/anie.196907811.
- Kenichi Fukui: The Role of Frontier Orbitals in Chemical Reactions (Nobel Lecture). In: Angewandte Chemie International Edition. Band 21, Nr. 11, 1982, S. 801–809, doi:10.1002/anie.198208013.
- James B. Hendrickson: Die Vielfalt thermischer pericyclischer Reaktionen. In: Angewandte Chemie. Band 86, Nr. 2, 1974, S. 71–100, doi:10.1002/ange.19740860203.
- Ulrich Lüning: Organische Reaktionen, 2. Auflage, Elsevier GmbH, München, 2007, S. 169, ISBN 978-3-8274-1834-0.