Aktivität (Chemie)
Die Aktivität ist eine thermodynamische Größe der Dimension Zahl, die in der physikalischen Chemie anstelle der Stoffkonzentration verwendet wird. Mit ihr gelten für reale Mischungen die gleichen Gesetzmäßigkeiten (bezüglich physikalischer Messparameter wie Gefrier-, Siedetemperatur, Leitfähigkeit von Elektrolytlösungen, elektrische Spannung, Dampfdrucken von Lösungsmitteln) wie mit der Konzentration für ideale Mischungen.

Der Aktivitätskoeffizient ist das Verhältnis der Aktivität zum Molenbruch mit der Einheit Eins.[1] Er liegt für ideales Verhalten, das bei unendlicher Verdünnung beobachtet wird, bei 1 und weicht mit zunehmender Konzentration ab. Die Aktivitätskoeffizienten können aber auch für Molaritäten und Molalitäten definiert werden.[2] Die Aktivität einer Spezies hängt mit der chemischen Zusammensetzung des restlichen Systems zusammen (beispielsweise über die Ionenstärke).

Einleitung
Mit Aktivitäten können Elektrolytlösungen (Leitfähigkeiten), Dampfdrücke eines Lösungsmittelsystems, Gasmengen in Flüssigkeiten, Gefrierpunkt-, Siedepunktänderungen, osmotische Drücke und auch Änderungen der Volumenmenge, der Temperaturveränderung von Lösungsmittelgemischen exakt beschrieben werden. Die tatsächliche Konzentration des zugesetzten Stoffes ergibt eine nicht proportionale Änderung der physikalischen Messgröße (z. B. Leitfähigkeit), so dass das Produkt aus Aktivitätskoeffizient und Konzentration die auf den beobachteten Messwert richtige Konzentration (die Aktivität) angibt. Die Abweichungen bezüglich der Messwerte basieren auf Coulombwechselwirkungen von Salzionen, Wasserstoffbrückenbindungen oder van der Waals-Wechselwirkungen in Lösungsmittelsystemen. Das Wort Aktivitätskoeffizient wurde erstmals von Svante Arrhenius zur exakten Beschreibung der Dissoziation gebraucht.[3]
In Elektrolytsystemen kann der jeweilige Aktivitätskoeffizient aufgrund der Ladungen einzelner Ionen berechnet werden oder recht schnell durch Leitfähigkeitsmessungen bestimmt werden. Der Aktivitätskoeffizient ist in Elektrolytlösungen fast immer kleiner als 1 und konvergiert bei sehr hoher Verdünnung gegen 1. Bei sehr konzentrierten Elektrolytlösungen kann der Aktivitätskoeffizient auch größer als 1 sein. Die Bestimmung eines individuellen Aktivitätskoeffizienten für eine Ionensorte ist nicht möglich, da Kationen und Anionen im Elektrolyten gemeinsam vorliegen. Für Elektrolytlösungen gibt man daher einen mittleren Aktivitätskoeffizienten an, der die physikalischen Abweichungen beider Ionen angibt. Ionen in Wasser und anderen Lösungsmitteln lagern sich an andere Ionen mit entgegengesetzter Ladung. Dadurch werden die Ionen abgeschirmt, es kommt zu Wechselwirkungen, und zwar umso stärker je höher die Konzentration und je höher die Ladung des betreffenden Ions ist. In sehr hoher Verdünnung (0,00001 molar) verschwinden diese Wechselwirkungen. Zur Bestimmung der Äquivalentleitfähigkeit aus der Grenzleitfähigkeit von Salzen, Säuren und Basen dürfen jedoch nicht die Aktivitätskoeffizienten der Debye-Hückel-Theorie verwendet werden. Die letzteren Koeffizienten gelten für das Massenwirkungsgesetz und für das Löslichkeitsprodukt, die Abhängigkeiten der Leitfähigkeit werden über das Kohlrauschsche Gesetz beschrieben.
Auch bei Mischungen von mehreren Lösungsmitteln kann es zu Wechselwirkungen kommen, die sich in Volumen-, Temperatur- oder Dampfdruckänderungen manifestieren. Für ein reines Lösungsmittel kann der Dampfdruck bei einer bestimmten Temperatur genau bestimmt werden. Daher sollte sich bei einem idealen Verhalten auch in einem Lösungsmittelgemisch in der Dampfphase die entsprechenden Gasmengen der Molenbrüche nach ihrem Anteilen im Lösungsmittelgemisch vorfinden (Daltonsches Gesetz). Bei vielen realen Lösungsmittelgemischen beobachtet man jedoch ein nichtideales Verhalten. Es können höhere oder geringere Dampfdrucke der Einzelkomponenten durch van der Waals Wechselwirkungen auftreten. Auch in diesem Falle verwendet man zur Beschreibung der realen Situation Aktivitätskoeffizienten. Dabei kann der Aktivitätskoeffizient auch größer als 1 werden. Bei der fraktionierten Destillation von Stoffgemischen ist daher die Kenntnis der Aktivitätskoeffizienten oder auch Fugazitätskoeffizienten (bei Gasdrücken) besonders wichtig, weil die in der Gasphase angereicherte Komponente so exakt bestimmt werden kann.
Auch beim umgekehrten Fall der Lösung eines Gases in einer Flüssigkeit ist die Gaslöslichkeit häufig annähernd proportional zum Gasdruck (Henrysches Gesetz). Auch in diesem Falle gibt es Abweichungen vom idealen Verhalten, so dass der Aktivitätskoeffizient benutzt wird. Ferner können beim Mischen von Lösungsmitteln oder beim Einfügen von Salzen in Lösungsmitteln Volumen-, Temperaturänderungen auftreten, die auch nicht proportional zur Konzentration des zugegebenen Stoffes sind.
Ideale Mischungen sind dadurch gekennzeichnet, dass keine Mischungseffekte auftreten, das heißt, dass sich das Volumen (bei konstantem Druck) und die Temperatur beim Mischen der reinen Komponenten nicht ändert und sich thermodynamisch wichtige Größen wie Enthalpie oder Wärmekapazität additiv aus den Werten der Einzelkomponenten multipliziert mit den jeweiligen Stoffmengenanteilen (Molenbrüchen) zusammensetzen. Dies ist nur der Fall, wenn sich die Komponenten chemisch sehr ähnlich sind, so dass die Wechselwirkungen zwischen Teilchen verschiedener Sorten gleich sind wie die Wechselwirkungen zwischen gleichen Teilchen. In der Regel wird dies nicht der Fall sein, so dass man es mit realen anstatt mit idealen Mischungen zu tun hat. In realen Mischungen sind die thermodynamischen Größen Volumen, Innere Energie und Enthalpie nicht mehr additiv aus den mit den Stoffmengenanteilen gewichteten Werten der Einzelkomponenten zusammengesetzt. Um die Gleichungen zur Beschreibung idealer Mischungen auch auf reale Mischungen anwenden zu können, wird die Aktivität eingeführt, die so festgelegt ist, dass bei dem Austausch der Stoffmengenanteile durch die Aktivitäten der additive Zusammenhang der thermodynamischen Größen für die Mischung aus den entsprechenden Größen für die Einzelkomponenten auch für reale Mischungen gegeben ist. In idealen Mischungen entspricht die Aktivität einer Komponente damit exakt dem Stoffmengenanteil der Komponente, in realen Mischungen werden die beiden Größen im Regelfall voneinander abweichen.
Definition
Die absolute Aktivität ist definiert durch:[4]
- .

Die relative Aktivität ist definiert als der Quotient der absoluten Aktivitäten des Zustandes und des Referenzzustandes:[5]

Hier steht für die absolute Temperatur, für das chemische Potential und für das chemische Potential des Referenzzustandes, typischerweise der Standardzustand (siehe Standardbedingungen).[6][7] ist die allgemeine Gaskonstante.




Bei der Verwendung des Begriffs Aktivität sollte stets angegeben werden, ob die relative oder absolute Aktivität gemeint ist.
Durch Einsetzen der Definition des chemischen Potentials , welche den Wechselwirkungsanteil enthält, erhält man:



Der Wechselwirkungsanteil des chemischen Potentials ist in idealen Systemen gleich Null.
In einer idealen Mischung ergibt sich das chemische Potential der Komponenten aus der Gleichung
- ,

wobei der Stoffmengenanteil (Molenbruch) der Komponente i ist.

Für eine reale Mischung muss eine Korrektur vorgenommen werden:

Die Aktivität ist mit dem Stoffmengenanteil durch folgende Beziehung verbunden:

Der dimensionslose Faktor wird als Aktivitätskoeffizient bezeichnet. Während in einer idealen Mischung keinerlei intermolekulare Wechselwirkungen vorhanden sind und somit die chemischen Potentiale und alle damit verbundenen Größen auf die Stoffmengenanteile der Komponenten zurückzuführen sind, sind in einer realen Mischung Wechselwirkung zwischen den Teilchen vorhanden. Diese Wechselwirkungen können z. B. elektrostatisch sein. Der Aktivitätskoeffizient beschreibt genau diese Abweichungen der Mischung vom Idealverhalten. Er wurde von Gilbert Newton Lewis 1907 als rein empirische Größe für starke Elektrolyte eingeführt. Svante Arrhenius und Jacobus Henricus van't Hoff gebrauchten den Ausdruck Aktivitätskoeffizient etwas zeitiger für den Dissoziationsgrad bei der Gefrierpunktserniedrigung durch Salze.[8]
Eine Herangehensweise ist, dass man sehr kleine Stoffmengen wählt. Dies führt (natürlich nur in der Theorie) zu folgenden Überlegungen/Ergebnissen:
- wenige Teilchen bedeutet einen großen Abstand der Teilchen und somit keine Wechselwirkungen
- es können Wechselwirkungen mit den Gefäßwänden vernachlässigt werden
Bei Lösungen wird noch angenommen, dass keinerlei nennenswerte Wechselwirkung zwischen Lösemittel und gelöstem Stoff vorhanden sind. Mathematisch lassen sich die Näherungen wie folgt ausdrücken:
- (allgemein)

- (in Lösungen)

- (in Gasgemischen)

Die Näherung für Lösungen ist bei Konzentrationen im Bereich von ca. relativ gut, wobei es in der Praxis auch auf die verwendete Messmethode ankommt. Der Divisor folgt aus der Dimension des chemischen Potentials: Soll dimensionslos sein, muss man die entsprechende Konzentration durch eine Konzentration teilen. Die Werte für c° und p° sind zwar beliebig, allgemein akzeptiert und von IUPAC seit 1982 als Standardbedingungen empfohlen sind die Standardkonzentration sowie der Standarddruck . Um nun wieder den Schritt zu realen Mischungen zu gehen, werden die benötigten Terme einfach mit dem Aktivitätskoeffizienten multipliziert.



Aktivität eines Lösungsmittels
In Lösungen ist die Aktivität eines Stoffes definiert über



wobei der Dampfdruck des reinen Lösungsmittels ist (der als Standardfugazität gewählt wird), der Dampfdruck des Lösungsmittels in der Lösung (eigentlich die Fugazität). a ist in diesem Fall analog zum Molenbruch zu sehen: Dies ist der Molenbruch, der bei Betrachtungen des Systems ausschlaggebend ist. Das Raoultsche Gesetz gilt für Lösungsmittel umso mehr, je näher der Molenbruch sich 1 annähert, d. h. je reiner das Lösungsmittel ist. Um das Verhalten anschaulich zu beschreiben, wurde hier ebenfalls ein Korrekturfaktor eingeführt, der nun wie folgt definiert:


mit für



Dieser Zusammenhang lässt sich aus dem chemischen Potential und dem Raoultschen Gesetz ableiten.
Grenzaktivitätskoeffizient
Der Grenzaktivitätskoeffizient ist der Aktivitätskoeffizient einer Komponente bei unendlicher Verdünnung () in einem Lösungsmittel oder einem Lösungsmittelgemisch.


Aktivität von Ionen und Elektrolyten
Die Bestimmung der Aktivitäten basierte auf der Dissoziationstheorie von Elektrolyten. Van't Hoff bestimmte Dampfdruckerniedrigung, osmotische Drücke, Siedepunktserhöhung, Gefrierpunktserniedrigung von Elektrolytlösungen und konnte bei sehr stark verdünnten Salzlösungen erhebliche Abweichungen zur Molgewichtsbestimmung feststellen.[9] Durch Leitfähigkeitsmessungen konnten fast identische Abweichungen wie bei der Gefrierpunktserniedrigung festgestellt werden. Später wurden Aktivitätskoeffizienten auch durch Potentialmessungen bestimmt. Zwischen den verschiedenen Methoden gibt es leichte Abweichungen zwischen den bestimmten Aktivitätskoeffizienten, so dass eine Angabe über die Nachweisart nicht fehlen sollte. Durch sehr preiswerte elektronische Messgeräte ist die Bestimmung von Aktivitätskoeffizienten in wässrigen Lösungen heute sehr einfach und unkompliziert geworden.
Um 1920 war es jedoch ein sehr mühsames Unterfangen für Chemiker aus Elektrolytlösungen die Aktivitätskoeffizienten zu bestimmen. Debye und Hückel (Debye-Hückel-Theorie) konnten im Jahr 1923 eine Theorie zum Verhalten von Elektrolyten in der Elektrolytlösung vorlegen, welche jedoch nur bei niedrigen Ionenstärken zuverlässige Werte liefert.[10]
Für verdünnte Elektrolyte lässt sich der Aktivitätskoeffizient aus der Debye-Hückel-Theorie berechnen. Dieses rein mathematische Verfahren ist jedoch nur bei Lösungen mit Ionenstärken unter 0,01 mol/l halbwegs genau. Dabei ist zu beachten, dass die oben angegebene Definitionsgleichung für jede individuelle Ionensorte einzeln gilt


Bei höheren Ionenstärken (I>0.1 M) kann die Davies-Gleichung zur Bestimmung der Aktivitätskoeffizienten eingesetzt werden: bei I=0,5 M ist der relative Fehler der Davies-Gleichung jedoch auch bereits 10 %[11]. Bei noch höheren Ionenstärken sind keine guten Näherungen bekannt und um verlässliche Werte für die Aktivitätskoeffizienten zu erhalten, müssen gE-Modelle eingesetzt werden.
Da jede Lösung elektroneutral sein muss, das heißt gleich viele positive wie negative Ladungen aufweisen muss, konnten individuelle Aktivitäten zwar nutzbringend berechnet, aber nicht gemessen werden. Diese vorherrschende Meinung geht bis in die 1920er Jahre auf Edward Guggenheim zurück. Damit wäre jedoch beispielsweise der pH-Wert nicht vollständig definiert, welcher die Aktivität von Wasserstoff-Ionen in Wasser beschreibt. In neuester Zeit, berichten einige Autoren einzelne Ionenaktivitäten experimentell gemessen zu haben[12]. Bevor einzelne Aktivitätskoeffizienten gemessen werden konnten, behalf man sich des Konzeptes der mittleren Aktivitätskoeffizienten.
Mittlerer Aktivitätskoeffizient
Für einen nach der Gleichung
- stöchiometrische Koeffizienten
- Ladungszahlen (negativ für Anionen)



vollständig dissoziierenden Elektrolyten gilt, wenn wir die Indizes „+“ für die Kationen und „-“ für die Anionen verwenden,



Zur Vereinfachung dieser Gleichung werden nun die mittlere Aktivität

der die stöchiometrischen Koeffizienten berücksichtigende Faktor

sowie der z. B. in galvanischen Zellen messbare mittlere Aktivitätskoeffizient

definiert. Für die mittlere Aktivität des Elektrolyten gilt dann mit als Konzentration des Elektrolyten


Beispiele:
- Für eine wässrige Lösung, die je 10−3 mol · dm−3 Kaliumchlorid (KCl) und Natriumsulfat (Na2SO4) enthält, wird aus dem Debye-Hückel-Grenzgesetz berechnet. Es ist , also



- Für eine 0,1-molare Magnesiumchlorid-Lösung (MgCl2) ist
- . Mit ergibt sich
- .



Aktivitätskoeffizient
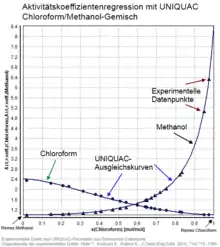
Ingenieurbereich
Im technischen Bereich werden für die Abschätzung des Aktivitätskoeffizienten sogenannte -Modelle wie zum Beispiel NRTL (Non-Random-Two-Liquid), UNIQUAC (Universal Quasichemical) und UNIFAC (Universal Quasichemical Functional Group Activity Coefficients) eingesetzt.

Elektrochemie
Nach der Interpretation von Debye und Hückel ist die elektrostatische potentielle Energie, die entsteht, wenn 1 Mol der Ionensorte A vom fiktiven ungeladenen Zustand auf seine reale Ladungsmenge innerhalb seiner entgegengesetzt geladenen Ionenwolke aufgeladen wird. Diese potentielle Energie ist somit negativ („Man muss Energie hineinstecken, um die Ionensorte A aus seiner Ionenwolke zu entfernen“).

Debye-Hückel:

Darin ist die universelle Gaskonstante und die Temperatur. W ist negativ; somit liegt für Systeme, die durch die Debye-Hückel Näherung beschrieben werden zwischen 0 und 1. Die Theorie von Debye-Hückel verlangt eine starke Verdünnung von A. Generell sind für beliebige Systeme aber auch Aktivitätskoeffizienten größer eins möglich.
Experimentelle Messungen ergaben eine Abhängigkeit zwischen der Ionenkonzentration von A und seinem Aktivitätskoeffizienten. Für große Konzentrationen von A wird größer als eins („Ich gewinne Energie, wenn ich A entferne“). Große Ionenkonzentrationen von A werden von der Debye-Hückel-Theorie nicht berücksichtigt.
Beispiel: Für 1-molare Essigsäure ist = 0,8 und für 0,1-molare Essigsäure 0,96.
Ein Verfahren zur Abschätzung von Aktivitätskoeffizienten ist die reguläre Lösungstheorie. Der Aktivitätskoeffizient wurde geschrieben a = f×c. Darin ist c die Konzentration, f der Aktivitätskoeffizient und a die Aktivität.
Literatur
- Gustav Kortüm: Lehrbuch der Elektrochemie, 5. Auflage, Verlag Chemie GmbH, Weinheim 1972.
- A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 81.–90. Auflage. Walter de Gruyter, Berlin 1976, ISBN 3-11-005962-2, S. 179–180.
Einzelnachweise
- Gerd Wedler: Lehrbuch der Physikalischen Chemie. John Wiley & Sons, 2012, ISBN 978-3-527-32909-0. S. 359.
- Walter J. Moore: Physikalische Chemie. Walter de Gruyter, 1986, ISBN 978-3-110-10979-5, S. 539.
- Zeitschrift f. Physik. Chemie I, 631 (1887)
- Eintrag zu absolute activity. In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.A00019 – Version: 2.3.2.
- Eintrag zu activity (relative activity). In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.A00115 – Version: 2.3.2.
- Eintrag zu standard state. In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.S05925 – Version: 2.3.2.
- Eintrag zu standard chemical potential. In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.S05908 – Version: 2.3.2.
- Max Le Blanc: Lehrbuch der Elektrochemie,Oskar Leiner Verlag Leipzig, 1922, S. 56.
- Gustav Kortüm: Lehrbuch der Elektrochemie, 5. Auflage, Verlag Chemie GmbH, Weinheim 1972, S. 146.
- P. Debye und E. Hückel: Physik Z., 24, 185 (1923).
- Ionic Equilibrium: Solubility and PH Calculations, ISBN 0471585262, Seite 49ff.
- Alan L. Rockwood: Meaning and Measurability of Single-Ion Activities, the Thermodynamic Foundations of pH, and the Gibbs Free Energy for the Transfer of Ions between Dissimilar Materials. In: ChemPhysChem. 16, 2015, S. 1978, doi:10.1002/cphc.201500044.