Massenspektrometrie
Massenspektrometrie bezeichnet ein Verfahren zum Messen der Masse von (historisch ursprünglich) Atomen oder (heute meist) Molekülen.
Die zu untersuchenden Moleküle werden dabei in die Gasphase überführt (Desorption) und ionisiert. Die Ionen werden anschließend durch ein elektrisches Feld beschleunigt und dem Analysator zugeführt, der sie nach ihrem Masse-zu-Ladung-Verhältnis m/z (auch m/q) „sortiert“, beispielsweise räumlich in Teilstrahlen auftrennt wie in einem Sektorfeld-Massenspektrometer. Die Moleküle können dabei fragmentiert werden.[1][2] Die Fragmentierung ist insbesondere bei den vergleichsweise komplexen Biopolymeren oftmals erwünscht, da die Fragmente leichter in die Gasphase überführt werden, beispielsweise bei der Untersuchung von Proteinen. Das für die Desorption in die Gasphase erforderliche Hochvakuum wird heute in der Regel durch den kombinierten Einsatz einer Drehschieberpumpe und einer Turbomolekularpumpe erzeugt.
Die Massenspektrometrie findet in vielen Bereichen Anwendung. Eingesetzt wird sie unter anderem bei der Charakterisierung von chemischen Verbindungen, in der Biochemie zur Untersuchung von Biomolekülen, in der medizinischen Chemie zur Identifizierung von Substanzen in Körperflüssigkeiten oder Organen, in kriminaltechnischen Untersuchungen, bei Dopingkontrollen, in der Umweltanalytik, in der Analytik von chemischen Kampfstoffen und Sprengstoffen. Es gibt sehr unterschiedliche Techniken, die sich je nach Aufwand, Anwendung und Genauigkeit unterscheiden. Vorteilhaft ist in vielen Bereichen, dass die Datenmenge recht gering ist und damit eine Kopplung mit Datenbanken von Massenspektren leicht möglich ist, z. B. Mascot für Proteine. Es ist auch verhältnismäßig leicht, ein Massenspektrometer mit einer HPLC-Anlage (meist ESI-MS) oder einem Gaschromatographen (oft EI-MS) zu koppeln und so nacheinander die verschiedenen Massenspektren der einzelnen Fraktionen zu erhalten.
Geschichte
.jpg.webp)
Die Massenspektrometrie basiert auf einer Hypothese, die vom britischen Chemiker William Prout im frühen 19. Jahrhundert aufgestellt wurde und besagt, dass jede Art von Atomen eine definierte Masse hat – damals als Atomgewicht bezeichnet. Er hatte festgestellt, dass die Masse der Atome einiger chemischer Elemente dem ganzzahligen Vielfachen der Masse des Wasserstoffatoms entsprach.[3][4] Spätere und genauere Messungen von Jöns Jakob Berzelius (1828) und Edward Turner (1832) schienen jedoch diese Hypothese zu widerlegen, denn es wurde z. B. für das Chlor-Atom eine Masse bestimmt, die das 35,45fache der Wasserstoffmasse beträgt. In der Mitte des 19. Jahrhunderts beobachtete Julius Plücker den Einfluss von magnetischen Feldern auf das Leuchten von Gasentladungsröhren.
Eugen Goldstein und Wilhelm Wien publizierten in den Jahren 1886 und 1898 die sogenannten Kanalstrahlen und ihre Ablenkung durch Felder. Sie hatten die weitreichenden Konsequenzen ihrer Entdeckungen jedoch 1886 noch nicht erkannt.[5][6][7][8][9][10]

Später, ab dem Jahr 1897, publizierte Joseph J. Thomson verschiedene Experimente,[11][9] in denen er in Vakuumröhren Kathodenstrahlen von verschiedenen Kathodenmetallen mit elektromagnetischen Feldern ablenkte, und stellte korrekte Gleichungen zum Zusammenhang zwischen Masse, Geschwindigkeit und Bahnradius auf. 1913 publizierte er eine Methode, um mit Hilfe eines Massenspektroskops Fotoplatten zu belichten und so qualitative und quantitative Untersuchungen an den in einer Röhre enthaltenen Gasen durchzuführen.
Im Jahr 1918 wurde von Arthur Jeffrey Dempster das erste moderne Massenspektrometer entworfen und gebaut, welches 100-fach genauer arbeitete als alle vorherigen Entwicklungen, und legte den Grundstein für das Design heutiger Massenspektrometer.[12] Es besaß einen magnetischen Sektoranalysator. Aufgrund dieser Entwicklung hat er im Jahr 1935 das Uranisotop mit der Masse 235 identifizieren können.
Ein Schüler von Thomson, der britische Chemiker und Physiker Francis William Aston, baute um dieselbe Zeit sein erstes Massenspektrometer, über das er 1919 berichtete.[13][14] Mit dessen neuer Technik konnte er die Isotope von Chlor (35Cl und 37Cl), von Brom (79Br und 81Br) und von Krypton (78Kr, 80Kr, 82Kr, 83Kr, 84Kr und 86Kr) beobachten. Aston wurde schließlich im Jahr 1922 mit dem Nobelpreis für Chemie für seine Untersuchungen der Isotope geehrt. Durch die Verwendung der Technik des Elektrofokussierens konnte er nicht weniger als 212 der damals bekannten 287 Isotope beobachten. Im Jahr 1932 entwickelte Kenneth Bainbridge ein Massenspektrometer mit einer Auflösung von 600 und einer Genauigkeit von 1:10.000.[15] Er verwendete es zur Bestätigung der Energie-Masse-Äquivalenz von Albert Einstein, E = mc2.[16]
1934 beschrieben Josef Mattauch und Richard Herzog ein doppelfokussierendes Massenspektrometer (Mattauch-Herzog-Geometrie, von Mattauch und Herzog 1936 gebaut), das Mattauch für die in der damaligen Zeit präzisesten Atommassenbestimmungen nutzte.
Im Jahr 1939 beschrieben Alfred Nier und Earl A. Gulbransen (1909–1992) das Isotopenverhältnis von Kohlenstoff.[17] Im Jahr 1946 wurde von William E. Stephens die gepulste Ionisation entwickelt, wodurch der messbare Massenbereich vergrößert wurde und die Grundlage für das erste Flugzeit-Massenspektrometer entstand.[18] Das erste Flugzeitmassenspektrometer wurde 1948 von A.E. Cameron und D.F. Eggers gebaut.[19] Eine deutliche Verbesserung der Auflösung wurde 1955 durch William C. Wiley und seinen Mitarbeiter Ian H. McLaren erzielt.[20]
Während des Manhattan-Projekts wurden zum Bau von Atombomben große Isotopenanreicherungsanlagen (Calutrons) gebaut, die nach dem Prinzip von Massenspektrometern arbeiteten.[21] In den 1950er Jahren wurde von Roland Gohlke und Fred McLafferty das erste Mal ein Massenspektrometer als Detektor für eine Chromatographie-Methode eingesetzt.[22][23][24] Beide koppelten einen Gaschromatographen mit einem Massenspektrometer. Durch diese Methode konnten das erste Mal Substanzgemische in einer Anlage getrennt und identifiziert werden.[24][25] Für die Anwendung in einer gaschromatographischen Methode müssen die entsprechenden Verbindungen jedoch im Vakuum flüchtig und dabei unzersetzt verdampfbar sein. Im Jahr 1953 entwickelte Wolfgang Paul den Quadrupol, der eine Selektion der Masse-Ladungsverhältnisse fliegender Ionen ermöglichte.[26]
Ebenso entwickelte Wolfgang Paul die Ionenfalle, mit der Ionen in einem definierten kleinen Raum gehalten werden konnten. Für seine Entdeckungen erhielt Wolfgang Paul 1989 den Nobelpreis für Physik. Ab 1959 wurde die Massenspektrometrie von Klaus Biemann und Kollegen zur Proteinidentifikation eingesetzt.[27]
Die bisherigen Methoden zur Erzeugung der benötigten Ionen waren sehr aggressiv und führten zu vielen Bruchstücken (Fragmente) beim Vermessen organischer Verbindungen. Daher setzte ab den 1960er Jahren die Entwicklung immer schonenderer Ionisationsmethoden ein. Mitte der 1960er Jahre wurde von Burnaby Munson und Frank H. Field die chemische Ionisation (CI) veröffentlicht.[28] Die Verbindung zweier Massenspektrometer über eine Kollisionskammer durch Jean Futrell und Dean Miller führte 1966 zur Entwicklung des ersten Tandem-Massenspektrometers.[29][30] Im Jahr 1969 publizierte H. D. Beckey die Felddesorption (FD).[31][32]
Im Jahr 1974 entwickelten Alan G. Marshall und Melvin B. Comisarow von der University of British Columbia inspiriert von den Fourier-Transformations-Kernspinresonanzspektroskopie- (FT-NMR) und Ionenzyklotronresonanz-Methoden (ICR) ein Fourier-Transform-Massenspektrometer (FT-ICR-Massenspektrometrie).[33] Die Unterscheidung von Radioisotopen und anderen Isotopen mit gleichem Masse-Ladungsverhältnis unter Verwendung eines Cyclotrons wurde erstmals von Richard A. Muller beschrieben.[34] Im Jahr 1977 wurde von Boris A. Mamyrin und Kollegen das Problem breiter initialer Energieverteilungen mit dem Reflektron gelöst.[35] In den späten 1970er Jahren verwendete Jim Morrison drei Quadrupole in Serie, bei denen der erste Quadrupol als Massenfilter, der Zweite zur Fragmentierung von Molekülen und der Dritte zur Detektion der Molekülionen diente. Durch Erhöhung des Drucks durch Zugabe eines Inertgases konnten Christie Enke, Richard Yost und Jim Morrison im Jahr 1979 eine kollisionsinduzierte Fragmentierung von zu untersuchenden Molekülen ohne eine Verwendung von Lasern erreichen.[36] Dadurch konnten auch Makromoleküle genauer untersucht werden. Im Jahr 1978 entwickelten Calvin Blakly, Mary McAdams und Marvin Vestal die Thermospray-Ionisation mit einer erhitzten Düse, durch die eine flüssige Probe mit Ammoniumacetat in ein Vakuum hinein verdampft wurde.[37] Ab 1981 wurde von Michael Barber und Kollegen die Ionisationsmethode des fast atom bombardment entwickelt, bei der die Ionisation durch beschleunigte Atome erreicht wurde.[38] Die Tandem-Massenspektrometrie wurde ab 1981 von Donald F. Hunt und Kollegen zur Proteinsequenzierung aus Proteingemischen eingesetzt.[39] Für die Elementaranalyse wurde ab 1980 von Robert S. Houk und Alan L. Gray die ICP-MS entwickelt, mit einer Sensitivität im Bereich von ppb oder ppt.[40]
Später erfolgten dann die weitere Entwicklung einer Vielzahl an Ionisationsmethoden für die unterschiedlichsten Zwecke wie zum Beispiel Elektrospray (ESI, ab 1968),[41][42] Chemische Ionisation bei Atmosphärendruck (APCI, ab 1974),[43] und Matrix-unterstützte Laser-Desorption/Ionisation (MALDI, ab 1985).[44][45]
Ab 1982 entwickelte John Bennett Fenn die Elektrospray-Ionisation für Biomoleküle.[46] Im Jahr 1987 veröffentlichte Koichi Tanaka eine flüssige Matrix mit Metallkolloiden für Biomoleküle.[47] Fenn und Tanaka erhielten im Jahr 2002 dafür den Nobelpreis für Chemie. Im Jahr 1999 entwickelte Alexander Makarov das Orbitrap-Massenspektrometer.[48]
Parameter eines Massenspektrometers
Ein Massenspektrometer wird durch verschiedene Parameter charakterisiert:[49][50][51] Die Massenauflösung, die Massengenauigkeit, der Massenbereich, der lineare dynamische Bereich und die Messrate.
Die Massenauflösung bezeichnet den minimalen Massenunterschied Δm, den zwei Ionen haben müssen, damit sie noch aufgelöst werden können. Die Auflösung eines Massenspektrometers wird in der Einheit Thomson (Th) angegeben, wobei aber trotzdem öfter nur das Auflösungsvermögen R angegeben wird. Dieses ist als Verhältnis einer Masse zum Massenunterschied der nächsten noch getrennt erscheinenden Masse (R = m/Δm) definiert. Zum Beispiel würde man bei einem Auflösungsvermögen von 4000 die Peaks bei 4000 Th und 4001 Th noch getrennt sehen, aber ebenso die Peaks bei 2000 Th und 2000,5 Th da 2000/(2000,5 − 2000) = 4000. In der Praxis werden die beiden Begriffe Auflösung und Auflösungsvermögen oft nicht exakt auseinandergehalten.
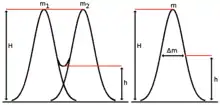
Es gibt verschiedene Definitionen der Auflösung:
- Bei der 10-%-Intensität-Methode definiert man Δm als die Massenabweichung, bei der die Intensität eines Peaks auf 10 % des Maximums absinkt.
- Bei der 10-%-Tal-Methode definiert man Δm als die Massenabweichung, bei der das Tal zwischen zwei gleich großen Peaks auf 10 % des Maximums absinkt.
- Bei der 50-%-Intensität-Methode definiert man Δm als die Massenabweichung, bei der die Intensität eines Peaks auf 50 % des Maximums absinkt.
- Bei der Halbwertsbreitenmethode (FWHM, full width at half maximum height) definiert man dm als die volle Peakbreite bei der halben Peakhöhe.
Die Massengenauigkeit gibt an, wie genau die Masse des Teilchens bestimmt werden kann. Diese Angabe erfolgt oft in parts per million (ppm), d. h. ein Molekül mit der nominellen Masse 500 kann bei einer Genauigkeit von 1 ppm auf 0,0005 u genau bestimmt werden.
Die Massenspanne ist der analysierbare Massenbereich eines Massenspektrometers. Der lineare dynamische Bereich ist der Bereich, bei dem die Signalintensität proportional zur Konzentration ist. Die Messrate ist die Anzahl an Messungen pro Zeiteinheit.
Aufbau eines Massenspektrometers
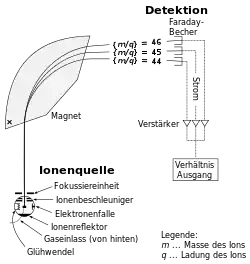
Ein Massenspektrometer (MS) besteht aus einer Ionenquelle, einem Analysator und einem Detektor. Jedes dieser Bauteile existiert in verschiedenen Bauformen und Funktionsprinzipien, die prinzipiell frei kombinierbar sind, obschon bevorzugte Kombinationen existieren. Diese werden im Folgenden beschrieben.
Ionenquelle
In der Ionenquelle wird der Analyt ionisiert. Dies kann mit Hilfe verschiedener Methoden erfolgen. Die Wahl der Methode ist hauptsächlich abhängig von der Art der zu analysierenden Substanz und davon, wie schonend ionisiert werden soll. Die Ionen werden meistens mit einem elektrischen Feld aus der Ionenquelle extrahiert und in den Analysator übergeben. Die Ionen können auf verschiedene Weisen erzeugt werden. Häufig kommen Stoßionisation, insbesondere Elektronenstoßionisation (EI) oder chemische Ionisation (CI), Photoionisation (PI), Feldionisation (FI), Fast Atom Bombardment (FAB), Inductively coupled plasma (ICP), Matrix-unterstützte Laser-Desorption/Ionisation (MALDI) und Elektrospray-Ionisation (ESI) vor.
Analysator
Im Analysator oder Massenselektor werden die Ionen nach ihrem Masse-zu-Ladung-Verhältnis getrennt (ist die Ladung bekannt, dann kann man damit direkt auf die Masse des Ions schließen). Es gibt mehrere sehr unterschiedliche Methoden, wie diese Massentrennung erfolgt. Abhängig von der Methode ist auch das Trennvermögen recht unterschiedlich. Die einzelnen Trennmethoden werden im Abschnitt Arten von Analysatoren behandelt.

Detektor
Der Detektor dient zur Erfassung der zuvor separierten Ionen. Als Detektor eingesetzt werden können Photomultiplier, Sekundärelektronenvervielfacher (SEV), Faraday-Auffänger, Daly-Detektoren, Mikrokanalplatten (MCP) oder Channeltrons. Der SEV wird teilweise in Kombination mit einer Konversionsdynode verwendet, bei der die Ionen aufgrund einer angelegten hohen Beschleunigungsspannung (bis zu 25 kV) auf eine Metalloberfläche prallen und der SEV dann die freiwerdenden Elektronen detektiert. In der Anfangszeit der Massenspektrometrie wurden auch Fotoplatten benutzt.
FT-ICR- und Orbitrap-Massenspektrometer messen Ströme (engl. image-currents), welche durch die sich bewegenden Ionenpakete in den Detektorplatten erzeugt werden. In diesem Fall werden die Ionen also nicht vom Detektor absorbiert und können deshalb mehrfach gemessen werden. Das trägt entscheidend zur hohen Messgenauigkeit dieser Instrumente bei.
Arten von Analysatoren
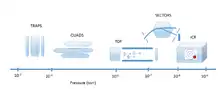
Massenspektrometer werden durch den jeweilig eingesetzten Analysator typisiert.
Einzelpartikel-Massenspektrometer
Mit einem Einzelpartikel-Massenspektrometer können Partikel in Echtzeit analysiert werden. Als Ergebnis werden Informationen über die chemische Zusammensetzung und Angaben über die Größe gewonnen. Das Instrument besteht aus vier Bestandteilen:
| Nummer | Bestandteil | Aufgabe |
|---|---|---|
| 1 | Einlasssystem | Aufnahme von Partikeln in eine Vakuumkammer |
| 2 | Detektionseinheit | Bestimmung der Fluggeschwindigkeit zur Feststellung der Partikelgröße und des Ablationszeitpunktes |
| 3 | Laserpuls | Verdampfung und Ionisierung von Partikeln |
| 4 | Massenspektrometer | Analyse des Masse-zu-Ladungsverhältnisses von Ionen |
Die dazugehörige Arbeitsmethode ist die Einzelpartikel-Massenspektrometrie. Die Partikel werden direkt aus der Umgebungsluft untersucht, was eine schnelle Analyse bei hoher Sensitivität ermöglicht. Weitere Vorteile sind eine hohe Zeitauflösung und eine niedrige Gefahr der Verunreinigung der Proben, da diese nicht erst zwischengelagert werden müssen. Auf diese Weise können exakte Statistiken aus großen Datenmengen erstellt werden. Die Analyse erfolgt mit Algorithmen auf der Basis von C++ und Matlab.[52][53]
Sektorfeld-Massenspektrometer

In Sektorfeld-Massenspektrometern werden die Ionen in statischen magnetischen Feldern oder zusätzlich statischen elektrischen Feldern abgelenkt.
Der Radius der Kreisbahnen, die sie in den Feldern durchlaufen, hängt von der Energie (im elektrischen Feld) und vom Impuls (im magnetischen Feld) der Ionen ab. In Kenntnis der Ladung, der Energie und des Impulses kann dann die Masse bestimmt werden. Sektorfeld-Massenspektrometer können so gebaut werden, dass Ionen mit leicht unterschiedlicher Geschwindigkeit auf einem Punkt im Detektor abgebildet werden (Geschwindigkeitsfokussierung). Auch Ionen, deren Flugbahn leicht geneigt ist, können auf einen Punkt abgebildet werden (Richtungsfokussierung). Massenspektrometer, die beides gleichzeitig können, nennt man doppelfokussierend. Die Fokussierung ist nötig, um bei hoher Auflösung noch eine akzeptable Intensität des Messsignals zu erhalten. Sektorfeld-Massenspektrometer erreichen Auflösungen von bis zu 100.000 und waren vor der Entwicklung der FT-Ionenfallen die Massenspektrometer mit der größten Auflösung. Heute werden sie nur noch selten verwendet, zum Beispiel in der Stabilisotopenmassenspektrometrie und in der Ultra-Spurenanalytik.
Quadrupol-Massenspektrometer
Im Quadrupol-Massenspektrometer werden die erzeugten Ionen durch ein statisches, elektrisches Feld beschleunigt und durchfliegen zentral vier parallel liegende Stabelektroden, deren Schnittpunkte mit einer Ebene senkrecht zur Zylinderachse ein Quadrat bilden (Quadrupol). Im Wechselfeld zwischen den Quadrupol-Stäben findet eine m/q-Selektierung statt, so dass jeweils nur Teilchen mit einer definierten Masse das Feld durchlaufen können.
Flugzeitmassenspektrometer (TOFMS)
Im Flugzeitmassenspektrometer (TOFMS) wird ausgenutzt, dass die Ionen beim Eintritt in den Analysator alle die gleiche Energie haben und leichte Ionen deshalb schneller sind als schwere. Daher erreichen beim Flug durch einen feldfreien Raum leichte Ionen den Detektor eher als schwere Ionen. Der Flugzeitanalysator besteht somit nur aus einem Rohr unter Vakuum mit einem sehr schnellen Detektor am Ende. Die Auflösung beträgt bis zu R = 15.000 (10-%-Methode). In der Praxis haben sich Geräte mit Ionenspiegeln bzw. Reflektron bewährt, bei denen die Flugstrecke durch ein zusätzliches elektrisches Feld am Ende der ursprünglichen Flugrichtung verdoppelt wird. Zusätzlich erreicht man durch diese Technik eine weitere Fokussierung.
Ionenfallen-Massenspektrometer

In Ionenfallen-Massenspektrometern werden die Ionen durch elektromagnetische Felder in einem definierten Bereich gehalten und können so analysiert und manipuliert werden. In der Quadrupol-Ionenfalle werden die Ionen durch ein Kühlgas, häufig Helium, gesammelt und stabilisiert. Dieses nimmt die thermische Energie der Ionen auf und sorgt dafür, dass sich die Ionen im Zentrum des Quadrupols sammeln und in einem ruhigen und geordneten Zustand vorliegen. Beim Anlegen einer bestimmten Spannung wird eine bestimmte Sorte an Ionen, die durch eine bestimmte Masse charakterisiert ist, instabil gemacht, die den Quadrupol verlässt und mittels eines Elektronenvervielfachers detektiert werden kann. In Ionenfallen-Massenspektrometern ist eine mehrfache Wiederholung von Anregung und Massenselektion möglich, ohne dass ein weiteres Bauteil benötigt wird. Folgende Typen von Ionenfallen-Massenspektrometer existieren:
- Quadrupol-Ionenfalle
- Linear trap
- Fouriertransformations-Ionenzyklotronresonanz (FT-ICR)
- Orbitrap
MS/MS (auch Tandem-Massenspektrometrie)

Um Fragmentierungen zu studieren oder auch um die Selektivität und Sensitivität (Nachweisgrenze) einer Quantifizierungsmethode entscheidend zu verbessern, koppelt man entweder mehrere Analysatoren hintereinander (sequentiell) oder arbeitet in Ionenfallen. Arbeiten die Geräte sequentiell, werden zwischen zwei Analysatoren sogenannte Kollisionszellen eingebaut, um den Ionen durch Stöße mit einem Inertgas (meist Stickstoff oder Argon) Energie zuzuführen. Daraufhin zerfallen die Ionen sehr spezifisch zu anderen (leichteren) Ionen.
Viele Kombinationen der Analysatoren sind denkbar. Die gängigsten sind Triple-Quadrupol (QqQ), Q-TOF, Tandem-TOF (TOF-TOF) und inzwischen auch als hochauflösende Massenspektrometrie als TRAP-FTICR und TRAP-Orbitrap.
Am weitesten verbreitet sind sogenannte Triple-Quadrupol-MS (QqQ, auch triple quads genannt), meist in der Kopplung mit HPLC. Dabei wird meist durch Elektrospray-Ionisation (ESI) ein Quasi-Molekülion produziert, welches im ersten Analysatorquadrupol isoliert und dann im zweiten Quadrupol – der sogenannten Kollisionszelle oder Stoßkammer – angeregt wird.
In die Stoßkammer kann ein Stoßgas (meist Argon, Helium oder Stickstoff) eingespeist werden. Dabei wird der Druck so gewählt, dass im Mittel ein erzeugtes Ion maximal einmal mit einem Gasmolekül kollidiert. Diese Methode ermöglicht es, erzeugte Ionen weiter zu fragmentieren.
Der dritte Quadrupol gibt die Möglichkeit zu „scannen“, also alle Produktionen des im ersten Quadrupol isolierten Ions (engl. parent ion) zu ermitteln oder selektiv nur ein bekanntes Fragmention zu beobachten. Durch das Erfassen aller Fragmentionen können Rückschlüsse auf die Struktur gezogen werden. Durch Beobachtung von nur ein oder zwei Fragmentionen kann sehr empfindlich und selektiv quantifiziert werden. Diese Technik wird auch als Multiple Reaction Monitoring (MRM) bezeichnet.
Daneben gibt es auch andere Techniken für MS/MS und auch sog. MSn, also Mehrfach-Massenspektrometrie. In Ionenfallen kann man ein Ion isolieren, und ihm dann entweder durch Kollision (meist mit Helium) oder auch durch Strahlung Energie zuführen und wiederum in der Trap fragmentieren (in-trap fragmentation). Dies kann mehrfach hintereinander durchgeführt werden (also MSn). Als Fragmentierungsmethoden neben der Kollision können hierzu auch Infrarotlaser, „Elektronenkanonen“ (engl. Electron Capture Dissociation) oder die Electron Transfer Dissociation (ETD) verwendet werden.
Triple-Quads sind im Bereich der HPLC-MS die meistgenutzten Massenspektrometer für quantitative Analysen. Die Tandem-Massenspektrometrie wird aber u. a. auch im Bereich der Proteincharakterisierung eingesetzt, z. B. in der De-Novo-Peptidsequenzierung.
Isobarenmarkierung
Bei der Isobarenmarkierung (englisch isobaric labeling)[54][55] werden die zu untersuchenden Moleküle mit unterschiedlichen Markierungen versehen,[55] die zwar isobar sind (die gleiche Ausgangsmasse besitzen), jedoch im Tandem-Massenspektrometer unterschiedlich schwere und somit unterscheidbare Fragmente (genauer Reporterionen) ergeben. Es gibt zwei kommerziell verfügbare Isobarenmarkierungen, Tandem Mass Tags (TMT)[54] und iTRAQ.[55] TMT existiert als Duplex oder 6-plex,[56] während iTRAQ als 4-plex oder 8-plex verfügbar ist.[57]
Kopplung mit Chromatographieverfahren
Bei sehr komplexen Proben (z. B. in der Lebensmittelanalytik) ist es nützlich, diese mit einem vorgelegten Trennverfahren aufzutrennen, bevor man sie dem Massenspektrometer zuführt. In diesem Sinn wird Massenspektrometrie oft zusammen mit Gaschromatographie (GC-MS) oder Flüssigchromatographie (LC-MS) betrieben. Weniger weit verbreitet sind die Kopplung mit Kapillarelektrophorese (CE-MS) und Ionenmobilitäts-Spektrometrie (IMS-MS). Teilweise werden auch mehrdimensionale Trenntechniken eingesetzt, z. B. GCxGC-MS. Flugzeitmassenspektrometer eignen sich besonders gut im Verbund mit mehrdimensionaler Gaschromatographie, weil damit sehr schnell Massenspektren über einen großen m/q-Bereich aufgenommen werden können (GCxGC-ToF-MS). Das GCxGC-Verfahren erlaubt eine genaue Auftrennung und Detektion verschiedener Verbindungsklassen aus komplexen Matrices (z. B. Erdölproben). Dafür werden zwei GC-Säulen mit unterschiedlicher Polarität hintereinander geschaltet.
Auswertung der Massenspektren
Voraussetzung für die Bestimmung der Masse m ist die Kenntnis der Ladung q des Ions, denn die Analysatoren können die Ionen nur nach dem Verhältnis m/q trennen. q ist jedoch immer ein ganzzahliges Vielfaches der Elementarladung e: q = z·e, und meistens ist z = +1 (einfach positiv geladen). Als Einheit von m/q wurde das Thomson Th vorgeschlagen: [m/q] = Th.
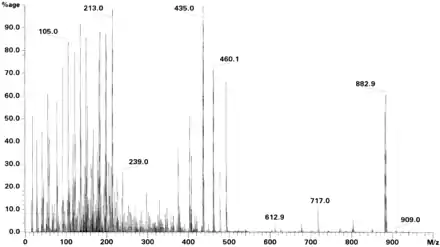
Die vom Detektor gelieferten Daten sind diskrete Werte, die normalerweise einen äquidistanten Abstand haben, welcher durch die Abtastung des Detektors vorgegeben ist. Diese Daten kann man direkt darstellen. Diese Darstellungsform nennt sich im Englischen „profile data“, welche von besonderem Interesse ist, falls die Peak-Breite wichtig ist. Alternativ dazu können die Daten weiter zu einem Histogramm verarbeitet werden, diese Darstellungsform nennt sich im Englischen „centroid data“: Den einzelnen Peaks wird entsprechend ihrer Fläche eine Intensität zugewiesen, welche sich am Ort des größten Wertes befindet.[58] Zunächst muss die Masse des Analyten bestimmt werden. Normalerweise ist das die Masse des schwersten detektierten Ions (Molekülpeak oder Molekülionpeak). Der Molekülionpeak gehört zum schwersten Ion, welches im Massenspektrum einer Substanz angezeigt wird, also dem einfach ionisierten Molekül. Allerdings wird bei der Elektronen-Ionisation oft ein Großteil der Ionen gespalten. Testweise kann die Elektronenenergie verringert werden, sodass weniger Ionen gespalten werden und der Molekülpeak deutlicher sichtbar wird.

Die weitere Auswertung basiert darauf, dass die Atome der verschiedenen chemischen Elemente einen unterschiedlichen Massendefekt haben. Daher kann aus einer sehr exakt bestimmten Masse eine Liste möglicher Summenformeln angegeben werden. Bei leichten Molekülen gibt es nur eine oder wenige passende Elementarzusammensetzungen. Mit steigender Masse oder zunehmender Anzahl an Heteroatomen steigt auch die Anzahl möglicher Kombinationen stark an.
Bei schwereren Molekülen stehen deshalb sehr viele mögliche Summenformeln zur Auswahl. Weitere Hinweise liefern die Isotopenzusammensetzungen der verschiedenen Elemente. So besteht der Kohlenstoff zum Beispiel zu 98,9 % aus 12C und zu 1,1 % aus 13C. Je nachdem, wie viele C-Atome im Molekül vorhanden sind, sind neben dem Hauptsignal im Spektrum Nebensignale zu finden, die vom Hauptpeak um 1 Th, 2 Th etc. entfernt sind und ein charakteristisches Intensitätsverhältnis zum Hauptsignal haben. Die Halogene Chlor und Brom, Schwefel und Silicium haben ebenfalls charakteristische Isotopenverhältnisse, die zur Identifizierung benutzt werden.
Die genannten Methoden sind auch auf die Bruchstücke anwendbar. Moleküle brechen oft an charakteristischen Stellen. Aus der Masse der Bruchstücke und evtl. weiteren Informationen kann schließlich die Strukturformel bestimmt werden.
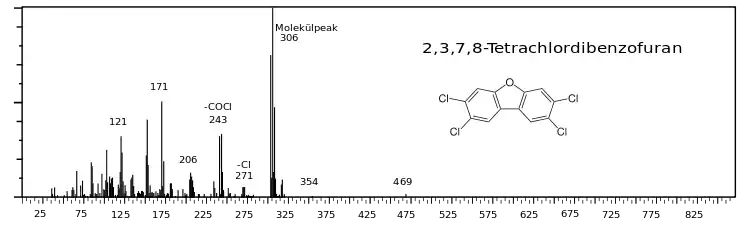
Dabei helfen vor allem bei mit positiver EI-Ionisierung erstellten Massenspektren auch Massenspektrenbibliotheken. Die bekanntesten sind unter den Kürzeln ihrer Vertreiber die Wiley- und die NIST-Massenspektrenbibliotheken. Durch den Peak Counting Score kann eine Identifizierung erfolgen.
Die Quantifizierung von Verbindungen wird bei der Massenspektrometrie dadurch erleichtert, dass bei der Analytik isotopenmarkierte (13C-markierte oder deuterierte) interne Standards verwendet werden können. (Isotopenverdünnungsanalyse)
Ein Problem bei der Datenauswertung stellen die proprietären Datenformate der einzelnen Gerätehersteller dar.[59] Die Daten werden in eigenen Binärdatenformaten vorgehalten. Meist werden vom jeweiligen Hersteller in die eigene Steuerungs- und Managementsoftware integrierte Auswertungsprogramme mitgeliefert. Um Programme von Dritten zu benutzen, bedarf es oft der Datenkonvertierung zum Datenexport, für die es im Bereich der Forschung frei erhältliche Lösungen gibt.[60]
Massenspektren bestehen aus mehreren unterschiedlichen Gruppen von Peaks:
- dem Molekülion
- Isotopenpeaks
- Fragmentpeaks
- metastabilen Peaks
Massenspektrometrie zeigt zunächst einmal einen Peak für das Molekülion, welches als Radikal-Kation M+. auftritt, als Resultat der Entfernung eines Elektrons aus dem Molekül. Der Molekülpeak ist jedoch nicht immer nachzuweisen oder kann sehr schwach ausgeprägt sein. In einer homologen Reihe verringert sich der Molekülpeak mit zunehmender Anzahl an Verzweigungen und mit zunehmender Masse. Das Molekülion zu identifizieren kann schwierig sein. Eine nützliche Hilfe ist dabei die Stickstoff-Regel: Wenn die Molekülmasse eine gerade Zahl ist, enthält die Verbindung keinen Stickstoff oder eine gerade Zahl an Stickstoffatomen. Molekülionen-Peaks sind häufig begleitet von einem M-1-Peak, der aus dem Verlust eines Wasserstoffradikals resultiert.
Weitere Peaks, mit einem m/z-Verhältnis größer als das des Molekülions, entstehen durch Isotopenverteilung. Der sogenannte M+1-Peak entsteht durch ein eingebautes Isotop höherer Masse, entweder 2H oder 13C; der M+2-Peak besitzt zwei Isotopen höherer Masse etc. Die natürliche Häufigkeit höherer Isotope ist für häufig vorkommende Elemente wie Wasserstoff, Kohlenstoff und Stickstoff gering und damit auch die Höhe der daraus resultierenden Isotopenpeaks, die Häufigkeit nimmt mit zunehmender Masse schnell ab. Bei den Halogenen Chlor und Brom dagegen sind höhere Isotope recht häufig, was sich in einem charakteristischen Signal äußert.
Peaks mit einer geringeren Masse als das Molekülion sind das Resultat aus Fragmentierungen. Der Peak mit der höchsten Intensität nennt sich Basispeak, er muss nicht unbedingt dem Molekülion entsprechen. Es existieren zahlreiche Reaktionswege für Fragmentierungen, aber lediglich neu gebildete Kationen tauchen im Massenspektrum auf, Radikalfragmente oder Neutralfragmente dagegen nicht.
Metastabile Peaks sind breite Peaks bei nicht-ganzzahligen Massewerten. Diese Peaks resultieren aus Fragmenten mit geringerer kinetischer Energie, wenn Fragmentierungen vor der Ionisationskammer stattfinden. Mit ihrer Hilfe lässt sich die Verwandtschaft zweier Peaks beweisen, die über einen einstufigen Zerfallsprozess verknüpft sind.
Anwendungen

Die Massenspektrometrie dient in der Analytik bzw. der analytischen Chemie als Analyseverfahren zur Bestimmung chemischer Elemente oder Verbindungen. In dieser Form werden Massenspektrometer in vielen Bereichen der Naturwissenschaften und der Technik für die Analyse von Materialien eingesetzt, unter anderem in der Chemie, Biologie, Archäologie und Klimatologie.
Auch in der Teilchenphysik werden Massenspektrometer verwendet. In diesem Bereich ist das Ziel jedoch weniger die Analyse von chemischen Elementen, sondern die Ermittlung der Massen von Elementarteilchen oder Atomkernen sowie der Detektion von noch unbekannten Teilchen.
Chemie
Für einen Analyten (die zu testende Substanz) wird die Häufigkeit, mit der geladene Moleküle (Ionen) und deren Massenfragmente auftreten, bestimmt. Die Massenspektrometrie ist eine wichtige Methode der analytischen Chemie bei der Aufklärung der Struktur und Zusammensetzung von Verbindungen und Gemischen, hier ist jedoch in der Regel eine angemessene Probenvorbereitung und eine Kopplung mit geeigneten gaschromatographischen oder liquidchromatographischen Trennverfahren erforderlich.[61] Der qualitative (Erkennung von unbekannten Substanzen) und quantitative (wie viel Substanz einer Verbindung ist vorhanden) Nachweis sehr kleiner Substanzmengen (ca. > 10−15 g = 1 fg (Femtogramm)) ist möglich.
Geologie
Isotopenverhältnisse verschiedener Elemente werden in der Geologie zur Altersdatierung von Gesteinskörpern sowie zur Thermochronologie verwendet und können Auskunft darüber geben, ob und wann ein Gestein nach seiner Entstehung noch einmal nachträglich erhitzt wurde. Sehr gut geeignet ist dafür unter anderem das Verhältnis von 39Ar zu 40Ar.
Archäologie
Isotopenverhältnisse einiger Elemente erlauben Rückschlüsse auf die Ernährung der Menschen, deren Knochen untersucht werden. Siehe auch Isotopenuntersuchung. Das Isotopenverhältnis von Kohlenstoff 14C zu 12C im organischen Material von archäologischen Funden erlaubt es, die Zeit seit der pflanzlichen Bildung der vermessenen Substanz zu ermitteln. Zur Messung von 14C wird die Beschleuniger-Massenspektrometrie herangezogen. Aus der Rekonstruktion „fossiler“ Proteine kann zudem auf die sie codierenden Gene und damit auf den Bau der DNA zurückgeschlossen werden.[62]
Biochemie
Massenspektrometrie wird in der Proteomik und Metabolomik verwendet, wo die Verwendung weitgehend jener in der Chemie entspricht. Biologische Proben, insbesondere Proteine, verlangen jedoch aufgrund der Molekülgröße und der speziellen Fragestellung (Identität, Sequenz, Posttranslationale Modifikation) bei der Aufklärung von systemischen Zusammenhängen eine besondere Probenvorbereitung und Messmethodik. Massenspektrometrische Methoden sind z. B. ITRAQ, ICAT, SILAC, Tandem Mass Tag oder die markierungsfreie massenspektrometrische Quantifizierung. Aminosäuresequenzen können durch De-Novo-Peptidsequenzierung bestimmt werden. Die Massenspektrometrie von Proteinen wurde von der Zeitschrift Nature Methods zur Methode des Jahres 2012 gekürt.[63]
Durch eine MassTag-PCR können UV-labil markierte Nukleinsäuren identifiziert und quantifiziert werden.
Klimatologie
Das Verhältnis der Häufigkeit bestimmter Isotope in Proben von Sedimenten, Baumringen und Eisbohrkernen lässt Rückschlüsse auf das Klima der Vergangenheit zu. Zum Beispiel verdampft Wasser, das das Isotop 16O enthält, leichter als solches, das das Isotop 18O enthält. Eiszeiten, bei denen große Mengen des Wassers als Eisschild dem Wasserkreislauf entzogen werden, verschieben die Häufigkeit dieser Isotope im Meer und damit auch im neu fallenden Schnee. Aus der Sauerstoff-Isotopenstufe kann auf die Menge des Inlandeises zu der Zeit geschlossen werden, als die Probe gebildet wurde.
Technik
Die Massenspektrometrie wird auch in vielen technischen Bereichen genutzt. Sie kann beispielsweise bei der Endpunkterkennung von Ätzprozessen eingesetzt werden. Ein anderer Anwendungsbereich ist die Einstellung und Optimierung der Gaszufuhr bei Beschichtungsprozessen (genauer chemische Gasphasenabscheidung). Hierbei wird das Abgas nach der Reaktion hinsichtlich unverbrauchter Reaktionsgase untersucht und die Gaszufuhr entsprechend angepasst. Die Massenspektrometrie kann aber auch zur Analyse der abgeschiedenen Materialien eingesetzt werden. Dabei können mithilfe von SIMS auch Tiefenprofile erstellt werden, was unter anderem bei der Analyse von dünnen Schichten eingesetzt wird.
Literatur
Allgemein
- Jürgen H. Gross: Massenspektrometrie - Ein Lehrbuch. Springer Verlag, Berlin/ Heidelberg, 2013, ISBN 978-3-8274-2980-3.
- Wolf Dieter Lehmann, Hans-Rolf Schulten: Physikalische Methoden in der Chemie: Allgemeine und Elektronenstoß-Massenspektrometrie I. In: Chemie in unserer Zeit. Band 10, Nr. 5, 1976, S. 147–158, doi:10.1002/ciuz.19760100504.
- Wolf Dieter Lehmann, Hans-Rolf Schulten: Physikalische Methoden in der Chemie: Massenspektrometrie II. In: Chemie in unserer Zeit. Band 10, Nr. 6, 1976, S. 163–174, doi:10.1002/ciuz.19760100602.
- Herbert Budzikiewicz, Mathias Schäfer: Massenspektrometrie – Eine Einführung. Wiley-VCH, Weinheim, 2005, ISBN 3-527-30822-9.
- Hans-Joachim Hübschmann: Handbook of GC/MS, Fundamentals and Applications. 3. Auflage. Wiley-VCH Verlagsgesellschaft, Weinheim, 2015, ISBN 978-3-527-33474-2. (auch als Online-Ressource verfügbar)
- Fred W. McLafferty, Frantisek Turecek (Deutsche Übersetzung): Interpretation von Massenspektren. 4. Auflage. Spektrum Akademischer Verlag, Heidelberg 1995, ISBN 0-935702-25-3.
- A. M. Boehm u. a.: Command Line Tool for Calculating Theoretical MS Spectra for Given Sequences. In: Bioinformatics. 20(16), 2004, S. 2889–2891. doi:10.1093/bioinformatics/bth328
- Alexander M. Lawson (Hrsg.): Mass Spectrometry - Clinical Biochemistry - Principles/Methods/Applications. Walter de Gruyter & Co., Berlin/ New York 1989, ISBN 3-11-007751-5.
- Mass Spectrometry Milestones. In: Nature Methods. (2015), Band 12, Supplement (PDF).
Spektrensammlungen
Spektrensammlungen liegen sowohl in gedruckten Werken als auch in gerätekompatiblen Dateiformaten vor. Letztere werden heute meist bereits mit den Geräten angeboten und für die komfortable Auswertung von unbekannten Massenspektren eingesetzt.
- M. Spiteller, G. Spiteller: Massenspektrensammlung von Lösungsmitteln, Verunreinigungen, Säulenbelegmaterialien und einfachen aliphatischen Verbindungen. Springer Verlag Wien/ New York 1973, ISBN 3-211-81117-6.
- A. Cornu, R. Massot: Compilation of Mass Spectral Data, Index de Spectres de Masse. Vol. 1 & Vol. 2, 2. Auflage. Heyden & Son, London/ Philadelphia/ Rheine 1979, ISBN 0-85501-086-X.
- K. Pfleger, H. Maurer, A. Weber: Mass Spectral and GC Data of Drugs, Poisons and Their Metabolites. Part I & II, VCH Verlagsgesellschaft, Weinheim 1985, ISBN 3-527-26303-9.
- NIST Standard Reference Database 1A, NIST/EPA/NIH Mass Spectral Library with Search Program: (Data Version: NIST 11, Software Version 2.0g) NIST-Spektrensammlung, auch unter Einschluss von Designerdrogen verfügbar.
- AIST Spectral Database for Organic Compounds SDBS, enthält außerdem noch 1H/13C-, FT-IR-, Raman- und ESR-Spektren.
ICP-MS
- W. Barger: Schulungsunterlagen zum ICP-MS Kurs 2006. LAS PerkinElmer (Germany) GmbH, Rodgau, unveröffentlicht.
- Robert S. Houk, Velmer A. Fassel, Gerald D. Flesch, Harry J. Svec, Alan L. Gray, Charles E. Taylor: Inductively Coupled Argon Plasma as an Ion Source for Mass Spectrometric Determination of Trace Elements. In: Analytical Chemistry. Band 52, Nr. 14, 1980, S. 2283–2289, doi:10.1021/ac50064a012.
- Simon M. Nelms (Hrsg.): Inductively Coupled Plasma Mass Spectrometry Handbook. Blackwell u. a., Oxford u. a. 2005, ISBN 0-8493-2381-9.
- Douglas A. Skoog, James J. Leary: Instrumentelle Analytik. Grundlagen, Geräte, Anwendung. Springer, Berlin u. a. 1996, ISBN 3-540-60450-2. (Übersetzung der 4. Auflage von Principles of Instrumental Analysis. Orlando 1992)
- Howard E. Taylor: Inductively Coupled Plasma-Mass Spectrometry. Practices and Techniques. Academic Press, San Diego CA u. a. 2001, ISBN 0-12-683865-8.
- Robert Thomas: Practical Guide to ICP-MS (= Practical Spectroscopy. 33). Dekker, New York NY u. a. 2004, ISBN 0-8247-5319-4.
Weblinks
- Linkkatalog zum Thema Massenspektrometrie bei curlie.org (ehemals DMOZ)
- Verzeichnis der ETH Zürich von Datenbanken und Nachschlagewerken mit MS-Spektren (Memento vom 12. März 2017 im Internet Archive)
- Simulation eines Massenspektrometers (deutsch; benötigt JavaScript)
- Geschichte der Massenspektrometrie (PDF; 210 kB)
- Massenspektrometrie – eine umfassende Darstellung der Theorie und der Technik
- Maturaarbeit von Gianluca Danieletto von der Kantonsschule Sargans: Animation der Massenspektrometrie
Einzelnachweise
- Eintrag zu mass spectroscopy. In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.M03748 – Version: 2.2.
- Eintrag zu mass spectrometry. In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.M03746 – Version: 2.2.
- William Prout: On the relation between the specific gravities of bodies in their gaseous state and the weights of their atoms. In: Annals of Philosophy. 6, 1816, S. 321–330 (online)
- William Prout: Correction of a mistake in the essay on the relation between the specific gravities of bodies in their gaseous state and the weights of their atoms. In: Annals of Philosophy. 7, 1816, S. 111–113 (online)
- E. Goldstein: Canalstrahlen. In: Sitzungsbericht der Preussischen Akademie der Wissenschaften. Band 691, 1886, S. 691–699.
- E. Rückardt: Zur Erinnerung an Wilhelm Wien bei der 25. Wiederkehr seines Todestages. In: Die Naturwissenschaften. 42. Jahrgang, Heft 3, 1955, S. 57–62 doi:10.1007/BF00589524.
- E. Rückhardt: Zur Entdeckung der Kanalstrahlen vor fünfzig Jahren. In: Die Naturwissenschaften. 24. Jahrgang, Heft 30, 1936, S. 465–467, doi:10.1007/BF01473963.
- J. J. Thomson: Cathode Rays. In: Philosophical Magazine. 44, 1897, S. 293, doi:10.1080/14786431003659214 (facsimile von Stephen Wright, Classical Scientific Papers, Physics, 1964 (Memento vom 30. Juli 2010 im Internet Archive)).
- J. J. Thomson: Rays of positive electricity. In: Proceeding of the Royal Society A. 89, 1913, S. 1–20 (Digitalisat auf JSTOR), wie exzerpiert in Henry A. Boorse, Lloyd Motz: The World of the Atom. Vol 1, 1966 (PDF) (Memento vom 17. November 2015 im Internet Archive)
- F. W. Aston: Kanalstrahlen und Atomphysik. In: Die Naturwissenschaften. 24. Jahrgang, Heft 30, 1936, S. 467–469, doi:10.1007/BF01473964.
- J.J. Thomson: XLII. . In: Philosophical Magazine Series 6. 19, 1910, S. 424, doi:10.1080/14786440308636816.
- A. J. Dempster: A New Method Of Positive Ray Analysis. In: Phys. Rev. Band 11, 1918, S. 316.
- F. W. Aston: LXXIV. A positive ray spectrograph. In: Philosophical Magazine Series 6. 38, 1919, S. 707, doi:10.1080/14786441208636004.
- Kevin M. Downard: Francis William Aston: The Man Behind the Mass Spectrograph. In: European Journal of Mass Spectrometry. 13, 2017, S. 177, doi:10.1255/ejms.878.
- Georges Audi: The history of nuclidic masses and of their evaluation. In: International Journal of Mass Spectrometry. Vol. 251, Nr. 2–3, 1. April 2006, S. 85–94, doi:10.1016/j.ijms.2006.01.048, arxiv:physics/0602050, bibcode:2006IJMSp.251...85A.
- Kenneth T. Bainbridge: The Equivalence of Mass and Energy. In: Phys. Rev. Vol. 44, Nr. 2, Juli 1933, S. 123, doi:10.1103/PhysRev.44.123.2, bibcode:1933PhRv...44..123B.
- Alfred O. Nier, Earl A. Gulbransen: Variations in the Relative Abundance of the Carbon Isotopes. In: Journal of the American Chemical Society. 61, 1939, S. 697, doi:10.1021/ja01872a047.
- William E. Stephens: A Pulsed Mass Spectrometer with Time Dispersion. In: Physical Review. 69, 1946, S. 674, doi:10.1103/PhysRev.69.674.2.
- A. E. Cameron, D. F. Eggers: An Ion “Velocitron”. In: Review of Scientific Instruments. 19, 1948, S. 605, doi:10.1063/1.1741336.
- William C. Wiley, Ian H. McLaren: Time-of-Flight Mass Spectrometer with Improved Resolution. In: Review of Scientific Instruments. 26, 1955, S. 1150, doi:10.1063/1.1715212.
- William E. Parkins: The Uranium Bomb, the Calutron, and the Space-Charge Problem. In: Physics Today. 58, 2005, S. 45, doi:10.1063/1.1995747.
- F. W. McLafferty: Mass Spectrometric Analysis Broad Applicability to Chemical Research. In: Analytical Chemistry. 28, 1956, S. 306, doi:10.1021/ac60111a005.
- F. W. McLafferty: Mass Spectrometric Analysis. Molecular Rearrangements. In: Analytical Chemistry. 31, 1959, S. 82, doi:10.1021/ac60145a015.
- R. S. Gohlke: Time-of-Flight Mass Spectrometry and Gas-Liquid Partition Chromatography. In: Analytical Chemistry. 31, 1959, S. 535, doi:10.1021/ac50164a024.
- Roland S. Gohlke, Fred W. McLafferty: Early gas chromatography/mass spectrometry. In: Journal of the American Society for Mass Spectrometry. Band 4, Nr. 5, Mai 1993, S. 367–371, doi:10.1016/1044-0305(93)85001-E.
- Wolfgang Paul, Helmut Steinwedel: Notizen: Ein neues Massenspektrometer ohne Magnetfeld. In: Zeitschrift für Naturforschung A. 8, 1953, S. 448–450 (online).
- K. Biemann, G. Gapp, J. Seibl: APPLICATION OF MASS SPECTROMETRY TO STRUCTURE PROBLEMS. I. AMINO ACID SEQUENCE IN PEPTIDES. In: Journal of the American Chemical Society. 81, 1959, S. 2274, doi:10.1021/ja01518a069.
- M. S. B. Munson, F. H. Field: Chemical Ionization Mass Spectrometry. I. General Introduction. In: Journal of the American Chemical Society. Band 88, Nr. 12, Juni 1966, S. 2621–2630, doi:10.1021/ja00964a001.
- Jean H. Futrell: Tandem Mass Spectrometer for Study of Ion-Molecule Reactions. In: Review of Scientific Instruments. 37, 1966, S. 1521, doi:10.1063/1.1720033.
- K.R. Jennings: Collision-induced decompositions of aromatic molecular ions. In: International Journal of Mass Spectrometry and Ion Physics. 1, 1968, S. 227, doi:10.1016/0020-7381(68)85002-8.
- H. D. Beckey: Field ionization mass spectrometry. In: Research/Development. 20(11),1969, S. 26.
- H. D. Beckey: Strukturbestimmung organischer Moleküle und quantitative Analysen mit dem Feldionisations-Massenspektrometer. In: Angewandte Chemie. 81, 1969, S. 17–18, 662.
- Melvin B. Comisarow, Alan G. Marshall: Fourier transform ion cyclotron resonance spectroscopy. In: Chemical Physics Letters. Band 25, Nr. 2, 15. März 1974, S. 282–283, doi:10.1016/0009-2614(74)89137-2.
- Richard A. Muller: Radioisotope dating with a cyclotron. In: Science. Band 196, Nummer 4289, April 1977, S. 489–494, doi:10.1126/science.196.4289.489, PMID 17837065. (PDF) (Memento vom 1. August 2010 im Internet Archive).
- B. A. Mamyrin, V. I. Karataev, D. V. Shmikk, V. A. Zagulin: The mass-reflectron, a new nonmagnetic time-of-flight mass spectrometer with high resolution. In: Soviet Physics JETP. (1973), Band 37, S. 45.
- R.A. Yost, C.G. Enke, D.C. McGilvery, D. Smith, J.D. Morrison: High efficiency collision-induced dissociation in an RF-only quadrupole. In: International Journal of Mass Spectrometry and Ion Physics. 30, 1979, S. 127, doi:10.1016/0020-7381(79)80090-X.
- C.R. Blakley, M.J. McAdams, M.L. Vestal: Crossed-beam liquid chromatoraph—mass spectrometer combination. In: Journal of Chromatography A. 158, 1978, S. 261, doi:10.1016/S0021-9673(00)89972-0.
- Michael Barber, Robert S. Bordoli, R. Donald Sedgwick, Andrew N. Tyler: Fast atom bombardment of solids (F.A.B.): a new ion source for mass spectrometry. In: Journal of the Chemical Society, Chemical Communications. 1981, S. 325, doi:10.1039/C39810000325.
- Donald F. Hunt, W. M. Bone, J. Shabanowitz, J. Rhodes, J. M. Ballard: Sequence analysis of oligopeptides by secondary ion/collision activated dissociation mass spectrometry. In: Analytical Chemistry. 53, 1981, S. 1704, doi:10.1021/ac00234a035.
- Robert S. Houk, Velmer A. Fassel, Gerald D. Flesch, Harry J. Svec, Alan L. Gray, Charles E. Taylor: Inductively coupled argon plasma as an ion source for mass spectrometric determination of trace elements. In: Analytical Chemistry. 52, 1980, S. 2283, doi:10.1021/ac50064a012.
- Malcolm Dole: Molecular Beams of Macroions. In: The Journal of Chemical Physics. 49, 1968, S. 2240, doi:10.1063/1.1670391.
- Masamichi Yamashita, John B. Fenn: Electrospray ion source. Another variation on the free-jet theme. In: The Journal of Physical Chemistry. 88, 1984, S. 4451, doi:10.1021/j150664a002.
- E.C. Horning, D.I. Carroll, I. Dzidic, K.D. Haegele, M.G. Horning, R.N. Stillwell: Liquid chromatograph—mass spectrometer—computer analytical systems. In: Journal of Chromatography A. 99, 1974, S. 13, doi:10.1016/S0021-9673(00)90841-0.
- Michael Karas, Doris Bachmann, Franz Hillenkamp: Influence of the wavelength in high-irradiance ultraviolet laser desorption mass spectrometry of organic molecules. In: Analytical Chemistry. 57, 1985, S. 2935, doi:10.1021/ac00291a042.
- M. Karas, D. Bachmann, U. Bahr, F. Hillenkamp: Matrix-assisted ultraviolet laser desorption of non-volatile compounds. In: International Journal of Mass Spectrometry and Ion Processes. 78, 1987, S. 53, doi:10.1016/0168-1176(87)87041-6.
- John B. Fenn, Matthias Mann, Chin Kai Meng, Shek Fu Wong, Craige M. Whitehouse: Electrospray ionization-principles and practice. In: Mass Spectrometry Reviews. 9, 1990, S. 37, doi:10.1002/mas.1280090103.
- Koichi Tanaka, Hiroaki Waki, Yutaka Ido, Satoshi Akita, Yoshikazu Yoshida, Tamio Yoshida, T. Matsuo: Protein and polymer analyses up tom/z 100 000 by laser ionization time-of-flight mass spectrometry. In: Rapid Communications in Mass Spectrometry. 2, 1988, S. 151, doi:10.1002/rcm.1290020802.
- Alexander Makarov: The Orbitrap: a novel high-performance electrostatic trap (Memento vom 21. März 2006 im Internet Archive).
- M. Mann, N. L. Kelleher: Precision proteomics: the case for high resolution and high mass accuracy. In: Proceedings of the National Academy of Sciences. Band 105, Nummer 47, November 2008, S. 18132–18138, doi:10.1073/pnas.0800788105. PMID 18818311. PMC 2587563 (freier Volltext).
- N. Tretyakova, M. Goggin, D. Sangaraju, G. Janis: Quantitation of DNA adducts by stable isotope dilution mass spectrometry. In: Chemical research in toxicology. Band 25, Nummer 10, Oktober 2012, S. 2007–2035, doi:10.1021/tx3002548. PMID 22827593. PMC 3495176 (freier Volltext).
- M. Scigelova, M. Hornshaw, A. Giannakopulos, A. Makarov: Fourier transform mass spectrometry. In: Molecular & cellular proteomics : MCP. Band 10, Nummer 7, Juli 2011, S. M111.009431, doi:10.1074/mcp.M111.009431. PMID 21742802. PMC 3134075 (freier Volltext).
- Anja Roth: Untersuchungen von Aerosolpartikel und Wolkenresidualpartikeln mittels Einzelpartikel-Massenspektrometrie und optischen Methoden, S. 11-17 – PDF-Datei, abgerufen am 9. August 2019
- Paul A. Baron, Klaus Willeke: Aerosol Measurement: Principles, Techniques, and Applications. 3. Auflage. John Wiley & Sons, New Jersey 2011, S. 130–132
- Thompson A, Schäfer J, Kuhn K et al.: Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. In: Anal. Chem.. 75, Nr. 8, 2003, S. 1895–904. doi:10.1021/ac0262560. PMID 12713048.
- Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ: Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. In: Mol. Cell. Proteomics. 3, Nr. 12, 2004, S. 1154–1169. doi:10.1074/mcp.M400129-MCP200. PMID 15385600.
- Dayon L, Hainard A, Licker V, Turck N, Kuhn K, Hochstrasser DF, Burkhard PR, Sanchez JC: Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. In: Anal. Chem.. 80, Nr. 8, 2008, S. 2921–31. doi:10.1021/ac702422x. PMID 18312001.
- Choe L, D'Ascenzo M, Relkin NR, Pappin D, Ross P, Williamson B, Guertin S, Pribil P, Lee KH: 8-plex quantitation of changes in cerebrospinal fluid protein expression in subjects undergoing intravenous immunoglobulin treatment for Alzheimer's disease. In: Proteomics. 7, Nr. 20, 2007, S. 3651–60. doi:10.1002/pmic.200700316. PMID 17880003.
- Jürgen H. Gross: Mass Spectrometry. Springer, 2011, ISBN 978-3-642-10709-2, S. 10, 11. (Google Books)
- A. M. Boehm u. a.: Extractor for ESI Quadrupole TOF Tandem MS Data Enabled for High Throughput Batch Processing. In: BMC Bioinformatics. 5. Jahrgang, 2004, S. 162, doi:10.1186/1471-2105-5-162.
- A. M. Böhm: Methoden zur effizienten Proteinidentifizierung anhand von Massenspektrometrie. Logos-Verlag, 2006.
- Joseph B. Lambert, Scott Gronert, Herbert F. Shurvell, David A. Lightner: Spektroskopie – Strukturaufklärung in der Organischen Chemie. 2. Auflage. Pearson, München 2012, ISBN 978-3-86894-146-3, S. 307–361.
- Jessica Hendy: Ancient protein analysis in archaeology. In: Science Advances. Band 7, Nr. 3, 2021, eabb9314, doi:10.1126/sciadv.abb9314.
- Anonym: Method of the Year 2012. In: Nature Methods. 10, 2012, S. 1, doi:10.1038/nmeth.2329.