Dünnschichtchromatographie
Die Dünnschichtchromatographie bzw. -chromatografie (DC oder TLC, englisch thin layer chromatography) ist ein physikalisch-chemisches Trennverfahren, das zur Untersuchung der Zusammensetzung von Proben genutzt wird. Besonders vorteilhaft bei dieser chromatographischen Methode ist der geringe apparative Aufwand, die Schnelligkeit, die hohe Trennleistung und der geringe Substanzbedarf. Eingesetzt wird sie zum Beispiel zum raschen Nachweis der Reinheit einer Substanz oder der Überprüfung der Identität mit einer Referenzsubstanz. Auch die Verfolgung des Reaktionsverlaufes von chemischen Umsetzungen im Labor ist mit wenig Aufwand möglich.


Geschichte
N. A. Izmailov und M. S. Shraiber, zwei russische Forscher, führten 1938 eine chromatographische Trennung mit einer horizontalen Dünnschichtplatte durch, auf die sie das Lösemittel auftropften. Doch ihre Methode wurde kaum beachtet. Erst als Justus G. Kirchner (1911–1987) am Fruit and Vegetable Laboratory des US-Landwirtschaftsministeriums in Südkalifornien und seine Mitarbeiter (darunter auch B. Harnischmacher) sich ab 1951 mit ihr befassten, wurde das Interesse anderer an der Methode geweckt. Zum Durchbruch verhalf ihr Egon Stahl, als er die Herstellung von leistungsfähigen Platten beschrieb. Von ihm stammt auch der Name Dünnschichtchromatographie.[1]
Theoretische Grundlagen
Grundprinzip der chromatographischen Trennung
Das Grundprinzip der Chromatographie gilt für alle chromatographischen Methoden und kann wie folgt kurz zusammengefasst werden: Zu analysierende Teilchen (Moleküle, Ionen) verteilen sich auf zwei Phasen in einem für die Teilchensorte charakteristischen Verhältnis. Wie das Verhältnis mit der Teilchensorte variiert, ist auch abhängig von den physikalischen Eigenschaften der beiden Phasen. Die Verhältnisse stellen sich als dynamische Gleichgewichte ein (Diffusion aufgrund der Wärmebewegung) und werden in der Chromatographie durch die Bewegung einer mobilen gegen eine stationäre Phase in Geschwindigkeitsunterschiede verwandelt. Die Geschwindigkeit einer Teilchensorte ergibt sich aus dem Produkt der Geschwindigkeit der mobilen Phase mit dem Zeitanteil, den die Teilchen in der mobilen Phase verbringen. Es wird angenommen, dass die Teilchen in der mobilen Phase (statistisch gesehen) dieselbe Geschwindigkeit haben wie die Laufmittelmoleküle. Sind sie an die stationäre Phase gebunden, ist die Geschwindigkeit gleich Null („stop and go“-Modell).
Bei geringen Verteilungsunterschieden noch eine Auftrennung zu erzielen, ist nicht nur eine Frage der Länge der Wegstrecke; entscheidend ist auch, dass die Teilchen sehr oft zwischen den Phasen wechseln. Dann gilt das Gesetz der großen Zahl und es entspricht der Zeitanteil, den das individuelle Teilchen in der mobilen bzw. der stationären Phase zubringt, genau dem Anteil der Teilchen dieser Sorte in den beiden Phasen.
Die DC zählt zu den flüssigchromatographischen Methoden. Damit lassen sich alle Proben, die ausreichend stabil sind und in Lösung gebracht werden können, untersuchen. Bei der DC wandert ein Lösungsmittel durch Kapillarkräfte in einem festen, feinporigen Trägermaterial (z. B. Kieselgel) aufwärts.
Stationäre Phase
Die stationäre Phase (Trennschicht) besteht aus einer dünnen Schicht eines sehr feinkörnigen Materials (z. B. Kieselgel, Kieselgur, Aluminiumoxid, Cellulose). Diese Trennschicht ist sehr gleichmäßig auf eine Trägerfolie oder Trägerplatte aus Kunststoff, Aluminiumblech oder Glas aufgetragen und kommerziell in unterschiedlichen Schichtdicken erhältlich. In der Regel kommt als stationäre Phase Kieselgel zum Einsatz (Normalphasenchromatographie), das aufgrund der freien endständigen Hydroxygruppen als polares Adsorbens für die Probenmoleküle dient. Der mittlere Porendurchmesser der Kieselgele beträgt meist 4 bis 100 nm, wobei der Porendurchmesser von 6 nm (Kieselgel 60, Merck) am gebräuchlichsten ist. Kieselgele enthalten Siloxan- oder Silanol-Gruppen.
Alternativ dazu kommen auch DC-Materialien mit anderen funktionellen Gruppen (z. B. Aminogruppen) zum Einsatz. Sie unterscheiden sich vom Standard-Kieselgel nicht nur in ihrer Polarität, sondern auch in der Basizität und führen damit zu völlig anderen Trennergebnissen. Auch oberflächenmodifizierte Kieselgele mit unpolaren Haftstellen (durch Kupplung mit Organochlorsilanen) werden eingesetzt (Umkehrphasenchromatographie, reversed phase). Die Reihenfolge, in der die verschiedenen Probemoleküle aufgetrennt werden, kehrt sich dann um – die polaren Moleküle laufen schneller, die unpolaren Moleküle werden stärker festgehalten. Vorteilhaft ist dabei unter anderem, dass auch sehr polare Proben untersucht werden können. Als weitere stationäre Phasen für die DC eignen sich auch Aluminiumoxid, Magnesiumsilikat, Kieselgur, Polyamid, Cellulose.
Die Trennung von geometrischen und Positions-Isomeren mit Doppelbindungen gelingt mittels Silbernitrat-Dünnschichtchromatographie[2]. Zur Trennung chiraler Proben werden reversed phase-DC-Platten eingesetzt, die mit dem Kupfer-Komplex eines chiralen Derivates[3] der Aminosäure L-Prolin beschichtet sind und die direkte dünnschichtchromatographische Trennung von Enantiomeren nach dem Prinzip der chiralen Ligandenaustauschchromatographie erlauben.[4][5][6]
Für spezielle Anwendungen kann auch das „Waschen“ der Platte vor dem Auftragen der Probe oder auch das Trocknen im Exsikkator oder Trockenschrank bei erhöhter Temperatur notwendig sein. Gewaschen werden die Platten durch Einstellen in eine Chromatographiekammer mit dem entsprechenden Lösungsmittel, bis die Fließmittelfront die Oberkante der Platte erreicht hat.
Mobile Phase
Als Laufmittel werden in der Normalphasen-DC unpolare organische Lösungsmittel in der Regel als Gemisch mit mäßig polaren Lösungsmitteln genutzt (z. B. Petrolether und Essigsäureethylester); in der Umkehrphasen-DC dagegen polare Laufmittel (z. B. Acetonitril und Wasser). Über das Mischungsverhältnis kann man die Polarität des Fließmittels steuern.
Die Adsorptionskapazität von Kieselgel für die funktionellen Gruppen nimmt in der Folge –COOH > –OH > –NH2 > –SH > –CHO > R2C=O > CO2R > –OCH3 > –HC=CH– ab.
Organische Carbonsäuren oder Alkohole weisen also auf Kieselgel eine sehr hohe Adsorption und damit geringe RF-Werte auf. Bei einem wenig polaren Laufmittel können diese Substanzen möglicherweise am Startfleck verbleiben.
Trennstrecke
Verschiedene Arten der Diffusion wirken einer guten Auftrennung entgegen. Entscheidend ist der rasche Wechsel der Teilchen zwischen den beiden Phasen. Daher ist es auch günstig, möglichst geringe Laufstrecken und feine, einheitliche Korngrößen des Schichtmaterials zu haben. Zu geringe und zu hohe Geschwindigkeiten der mobilen Phase wirken sich negativ aus. Eine zu geringe Geschwindigkeit begünstigt eine Vergrößerung der Zonen, in denen sich die Probemoleküle aufhalten. Je mehr Zeit zur Verfügung steht, desto größer ist die Rolle, die Diffusionsprozesse innerhalb der mobilen Phase spielen. Ist die Geschwindigkeit zu hoch, kommt es seltener zu einem Wechsel der Teilchen zwischen der mobilen und der stationären Phase. Das führt zu einer größeren statistischen Streuung und ist ebenfalls unerwünscht. Bei allen chromatographischen Methoden gibt es entsprechend eine optimale Geschwindigkeit der mobilen Phase, die durch die Van-Deemter-Gleichung beschrieben wird. Je feiner die Korngrößen sind (bzw. die Dimensionen), desto höher kann die Geschwindigkeit werden. Daraus folgen auch ökonomische Vorteile.
Bei der DC ist die erwünschte räumliche Auftrennung zwischen den verschiedenen Probekomponenten der gesamten Laufstrecke proportional (Abstand Startlinie – Laufmittelfront). Die Vergrößerung der einzelnen Zonen auf Grund statistischer Effekte ist geringer (nicht der Laufstrecke proportional, sondern der Wurzel der Laufstrecke). Daher ist es bei schwierigen Trennungen sinnvoll, größere DC-Folien und Laufstrecken zu verwenden.
Mit der Dünnschichtchromatographie lässt sich auf einer 15 cm langen Laufstrecke eine Trennleistung von ca. 400–3000 theoretischen Böden erzielen.
Praktische Durchführung
Probenauftrag

Die zu untersuchende Substanz wird in einem geeigneten Lösungsmittel gelöst und mit Hilfe einer Kapillare punkt- oder strichförmig aufgetragen. Dies geschieht bei der eindimensionalen DC auf der Startlinie der Folie oder Platte, bei der zweidimensionalen DC (siehe unten) in einer Ecke. Die zu trennende Substanzmenge beträgt ca. 5–20 Mikrogramm. Es ist entscheidend, die Auftragzonen möglichst eng zu halten (wenige Millimeter). Kommerziell erhältlich sind auch DC-Folien mit so genannten Konzentrationszonen unterhalb der Startlinie, beschichtet mit einem Material besonders geringer Adsorption. Die Substanzen in den dort womöglich ungenau platzierten oder großen Probeflecken lösen sich dann unmittelbar bei Durchgang der Lösemittelfront und erreichen mit ihr die Startlinie, in Laufrichtung zusammengestaucht. Für eine besonders gleichmäßige, quantitativ reproduzierbare Auftragung stehen auch Maschinen zur Verfügung, welche die Lösung mit Hilfe von Druckluft oder Stickstoff aufsprühen. Nach dem Auftragen muss die Platte getrocknet werden, da restliches Lösungsmittel das Ergebnis verändern kann.
Neben den Proben werden auf der Startlinie in vielen Fällen auch Lösungen von reinen Vergleichssubstanzen oder Vergleichsmischungen aufgetragen.
Auftrennung
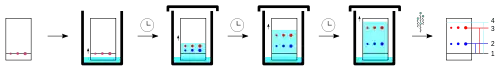
Nach dem Auftragen wird die Platte senkrecht in eine Chromatographiekammer mit einem geeigneten Fließmittel (mobile Phase) eingestellt. Um eine Beeinflussung der Ergebnisse (zu große ) durch das Verdampfen des Fließmittels zu verhindern, führt man die Trennung in einer mit dem Fließmittel gesättigten Atmosphäre in einem geschlossenen Gefäß durch. Zur besseren Sättigung des Dampfraumes mit Laufmittel kann ein Filterpapier eingelegt werden.

Das Fließmittel saugt sich nun über Kapillarkräfte in die stationäre Phase nach oben. Sobald die Flüssigkeit die Startlinie erreicht hat, lösen sich die Substanzen in ihr. Die Moleküle sind nun den Anziehungskräften der stationären Phase einerseits und den Anziehungskräften der mobilen Phase andererseits ausgesetzt. Je nach Kräfteverhältnis bleibt ein Teilchen eher am Startpunkt oder es wandert mit der mobilen Phase nach oben. Je unpolarer das Fließmittel und je polarer eine Substanz, desto weniger wandert die Substanz. Die Lösungsmittelpolarität ist dabei analog wie in der Säulenchromatographie.
Kurz bevor die Laufmittelfront das obere Ende der Platte erreicht, wird die Platte aus der Chromatographiekammer entnommen und möglichst zügig getrocknet.
Auswertung
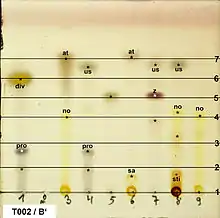
Im einfachsten Fall sind die getrennten Substanzen beim Betrachten unter UV-Licht als Punkte sichtbar. Alternativ können sie vor der Chromatographie mit Chromophoren derivatisiert werden, damit sie UV-aktiv werden. Auch das Besprühen mit oder Tauchen in Reagenzienlösungen sind weitere Möglichkeiten.
Viele Schichtmaterialien enthalten Zusätze, die im UV-Licht fluoreszieren und an denjenigen Stellen dunkle Fluoreszenzlöschung zeigen, an denen sich die getrennten Stoffe befinden. Diese Fluoreszenzfarbstoffe dürfen während der chromatographischen Trennung nicht wandern. Gebräuchlich sind vor allem Mangan aktiviertes Zinksilikat (mit UV-Licht der Wellenlänge 254 nm bestrahlt) und Calciumwolframat (mit UV-Licht der Wellenlänge 366 nm bestrahlt). Tatsächlich handelt es sich bei der Methode nicht um eine Fluoreszenzlöschung im engeren Sinne. Probemoleküle werden sichtbar, wenn sie im Bereich von 254 nm oder 366 nm UV-Licht absorbieren. Es gelangt dann weniger UV-Licht zu den Fluoreszenzfarbstoffmolekülen (dunkle Flecken auf grün bzw. blau leuchtendem Hintergrund zu sehen). Dazu müssen genügend viele funktionelle Gruppen bzw. genügend große Systeme mit konjugierten Doppelbindungen vorhanden sein. Gesättigte Kohlenwasserstoffe und viele Aminosäuren sind daher mit dieser Methode nicht nachzuweisen, aromatische Verbindungen z. B. sehr leicht bei 254 nm.
Auch die Eigenfluoreszenz bestimmter Stoffe oder andere Eigenschaften wie Radioaktivität können zur Detektion herangezogen werden.
Bei der Verwendung von Sprüh- oder Tauchreagenzien[7][8] (z. B. NBD-Cl, Molybdatophosphorsäure oder 2,7-Dichlorfluorescein) laufen Farbreaktionen ab, die empfindlich und spezifisch genug sind, um zum Nachweis bestimmter funktioneller Gruppen verwendet zu werden. Über die Auswahl der Farbreaktion lässt sich der Informationsgehalt bei der DC wesentlich erhöhen. Alternativ werden Reaktionen eingesetzt, die allgemein wirksam sind (zum Beispiel Oxidation mit Hilfe von Salpetersäurelösungen oder Ioddampf). Bei einer Reihe von Farbreaktionen ist es erforderlich, die Folie nach dem Besprühen bzw. der Tauchung zu erhitzen.
Eine weitere, sehr einfache Methode besteht in der Bedampfung mit molekularem Iod. Dazu genügt es, in ein Glasgefäß ein paar Iod-Kristalle zu legen. Sie sublimieren, das heißt, sie verdampfen direkt bei Raumtemperatur, bilden einen violetten Dampf von Diiodmolekülen. Durch Einlegen einer DC-Folie in einen solchen Trog werden über Diffusion und Reaktion mit den Molekülen der Substanzflecken binnen kurzer Zeit lockere Komplexverbindungen gebildet (lila oder braun). Vorteil bzw. Nachteil der Methode: die Iod-Verbindungen zerfallen relativ rasch.
In der Biochemie ist eine saure Ninhydrin-Lösung ein häufiges Sprühreagenz, um Aminosäuren zu detektieren. Hierbei wird das Ninhydrin über die Schiffsche Base und durch eine Decarboxylierung sowie Hydrolyse zu Ruhemans Violett. Durch Auftragen von Referenzproben, die unter gleichen Bedingungen gleich weit wandern wie entsprechende Probekomponenten, kann man das qualitative Auftreten von Stoffen nachweisen. Hierzu wird die Lage der verschiedenen Punkte mit der Lage der Referenzproben verglichen.
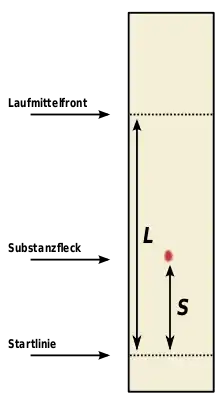
Um verschiedene DC vergleichen zu können, werden die so genannten -Werte (Retentionsfaktor, Rückhaltefaktor, ratio of fronts) berechnet. Es handelt sich dabei um das Verhältnis von Laufstrecke der Substanz () zur Laufstrecke des Laufmittels (): . Die -Werte sind bei gleichem Plattenmaterial und gleicher Laufmittelzusammensetzung Stoffkonstanten. Auch Anwendung findet der -Wert, wobei die Laufstrecke des Substanzflecks im Verhältnis zu der Laufstrecke eines Standards gesetzt wird. Der Standard ist dabei meist ein Reinstoff. Anhand des -Werts ist also eine qualitative Auswertung möglich.





Die quantitative Auswertung kann mit einem Densitometer ausgeführt werden. Diese Geräte bieten als sogenannte TLC-Scanner die Möglichkeit der Messung im sichtbaren und im ultravioletten Spektralbereich. Sie können auch zur Messung der Fluoreszenz eingesetzt werden. Auch reguläre Flachbettscanner können für eine densiometrische Auswertung in der DC genutzt werden.[9]
Neue Geräteentwicklungen ermöglichen auch die direkte Kopplung der Dünnschichtchromatographie mit der Massenspektrometrie.[10] So kann auch eine zuverlässige Identifizierung der chromatographisch getrennten Komponenten über deren Massenspektrum durchgeführt werden.
Komplexere Techniken und Methoden
Neben der bisher beschriebenen linearen und eindimensionalen Dünnschichtchromatographie sind für spezielle Anwendungen und Trennaufgaben Techniken entwickelt worden, um beispielsweise komplexere Substanzgemische auftrennen zu können. Die Hochleistungsdünnschichtchromatographie stellt eine solche Weiterentwicklung dar.
Zweidimensionale DC
Bei der zweidimensionalen DC wird nach der ersten Entwicklung das Laufmittel abgedampft, die Platte um 90° gedreht und – in der Regel in einem anderen Laufmittel – eine zweite Entwicklung durchgeführt. Dadurch kann eine bessere Auftrennung bei Multikomponentengemischen erreicht werden. Die Identifizierung ist aber aufwendiger, da keine Referenzsubstanzen mitlaufen können. Vor der zweiten Entwicklung kann die DC-Platte auch bearbeitet werden (zum Beispiel Bestrahlung mit UV-Licht) bevor die zweite Entwicklung im gleichen Fließmittel stattfindet (Transport-Reaktion-Transport-Technik, kurz TRT-Technik)
Zirkulare Dünnschichtchromatographie
Eine alternative Technik zur linearen DC stellt die so genannte zirkulare Dünnschichtchromatographie (abgek. CLC vom engl. Centrifugal Layer Chromatography oder RPC von Rotary Planar Chromatography) dar.[11] Hierbei werden runde Glasscheiben verwendet, die ringförmig mit der stationären Phase beschichtet sind. Die Scheibe wird mit Hilfe eines Elektromotors in rasche und gleichmäßig kontrollierte Rotation versetzt. Die Probelösung wird mit Hilfe einer Pumpe zum inneren Rand der Schicht geleitet, vorher und nachher das entsprechende Fließmittel.
Da die stationäre Phase in der Regel einen Farbstoff enthält, der bei UV-Licht der Wellenlänge 254 nm oder 366 nm fluoresziert, kann der Trennvorgang durch Bestrahlung mit einer UV-Lampe kontrolliert werden. Am Anfang des Trennprozesses befindet sich die Probe in einer wenige Millimeter starken Kreiszone am inneren Rand der Scheibe. Mit fortschreitender Trennung wird das Substanzgemisch der Probe in eine Reihe von Ringen aufgespalten, die getrieben von der Fliehkraft nach außen wandern.
Präparative Dünnschichtchromatographie
Die DC kann auch präparativ, d. h. zur Reinigung kleiner Substanzmengen genutzt werden. Dann wird sie auch PLC (engl. für Preparative Layer Chromatography) genannt. Hierbei werden in der Regel auf Glasplatten mit dickeren stationären Phasen (bis zu 2 mm) größere Mengen (bis zu 100 mg) der zu trennenden Substanzmischung strichförmig aufgetragen. Nach dem Trennlauf befinden sich die voneinander getrennten Substanzen als Linien in verschiedenen Höhen. Die Zielsubstanz kann dann mitsamt dem Trägermaterial mechanisch abgeschabt werden. Durch einfache Filtration mit einem geeigneten Lösungsmittel wird sie von der stationären Phase eluiert und so rein erhalten.
Auch von den üblichen (analytischen) DC-Folien mit dünner stationärer Phase lassen sich genügende Mengen Reinsubstanz erhalten, um sie für empfindliche Analyseverfahren wie die Massenspektrometrie oder die Infrarotspektroskopie nutzen zu können.
Die zirkulare DC kann ebenfalls präparativ genutzt werden. Hierbei wird die gewünschte Probenkomponente, nachdem sie am äußeren Rand der Trennschicht angelangt ist, gemeinsam mit dem Laufmittel in einem entsprechenden Sammelbehälter aufgefangen.
Um größere Substanzmengen bei weit geringerem apparativen Aufwand zu reinigen, nutzt man heute eher säulenchromatographische Techniken wie die Flash-Chromatographie.
Vorteile und Nachteile der Dünnschichtchromatographie
Im Gegensatz zu den leistungsfähigeren Chromatographie-Verfahren wie Gaschromatographie und Hochleistungsflüssigkeitschromatographie kommt die DC mit geringem apparativen Aufwand aus und stellt sich als schnelles, vielseitiges und preiswertes Analyseverfahren dar.
Die Gaschromatographie lässt sich nur bei Proben anwenden, die unzersetzt verdampfbar sind. Bei der Flüssigchromatographie gibt es wenig Einschränkungen. Fast immer lässt sich ein Weg finden, eine Probe aufzulösen. Im Vergleich zu den säulenchromatographischen Methoden besteht bei der Dünnschichtchromatographie der Vorteil, dass Proben, die Gruppen von Komponenten enthalten, die sich jeweils stark in der Polarität unterscheiden, leichter zu erfassen sind. Laufmittelwechsel ist nicht so leicht möglich wie bei der Säulenchromatographie. Es ist aber möglich, zunächst in einem Laufmittel zu entwickeln und nach Zwischentrocknung in einem anderen (das sich in der Polarität stark unterscheidet).
Nachteilig ist bei der analytischen Anwendung der DC, dass es schwerer ist, eine quantitative Analyse durchzuführen. Bei bestimmten Aufgabenstellungen genügt es allerdings schon, die Mengenverhältnisse abzuschätzen (Fortschritt einer chemischen Reaktion). Die früheren Probleme einer zuverlässigen Quantifizierung konnten in den letzten Jahren durch Entwicklung leistungsfähiger Densitometer – wie oben erwähnt – überwunden werden. Die Qualitätskriterien der quantitativen Auswertung genügen inzwischen den Richtlinien für die Gute Laborpraxis.
Literatur
- H.-P. Frey, K. Zieloff: Qualitative und quantitative Dünnschichtchromatographie. VCH, Weinheim 1993, ISBN 3-527-28373-0.
- F. Geiss: Die Parameter der Dünnschichtchromatographie. Vieweg, Braunschweig 1972, ISBN 3-528-08299-2.
- Elke Hahn-Deinstrop: Dünnschicht-Chromatographie-Praktische Durchführung und Fehlervermeidung. Wiley-VCH Verlag, Weinheim 1998, ISBN 3-527-28873-2.
- Justus G. Kirchner: Thin-layer chromatography. 2. Auflage. Wiley, New York 1978, ISBN 0-471-93264-7.
- Peter Pachaly: Dünnschichtchromatographie in der Apotheke, Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart 1982, ISBN 3-8047-0624-X
- Joseph Sherma, Bernard Fried (Hrsg.): Handbook of Thin-Layer Chromatography (= Chromatographic Science. Band 55). Marcel Dekker, New York NY u. a. 1991, ISBN 0-8247-8335-2.
- Lloyd R. Snyder, Joseph H. Kirkland, John W. Dolan: Introduction to Modern Liquid Chromatography. 3. Auflage. Wiley-Interscience, Hoboken NJ 2010, ISBN 978-0-470-16754-0.
- Egon Stahl (Hrsg.): Dünnschicht-Chromatographie: Ein Laboratoriumshandbuch. Springer, Berlin u. a. 1962.
- Felix Schumm: Dünnschichtchromatogramme – auch für den Amateur möglich. In: Aktuelle Lichenologische Mitteilungen. Nr. 9, 2002, S. 8–22.
- Colin Poole: Instrumental Thin-Layer Chromatography. Elsevier, Oxford 2014, ISBN 978-0-12-417223-4.
Weblinks
Einzelnachweise
- Joseph C. Touchstone: Practice of Thin Layer Chromatography. 3. Auflage. Wiley, New York NY u. a. 1992, ISBN 0-471-61222-7, S. 3–4.
- Beate Breuer, Thomas Stuhlfauth, Heinrich P. Fock: Separation of fatty acids or methyl esters including positional and geometric isomers by alumina argentation thin-layer chromatography. In: Journal of Chromatographic Science. Band 25, Nr. 7, 1987, ISSN 0021-9665, S. 302–306, doi:10.1093/chromsci/25.7.302.
- Kurt Günther, Jürgen Martens, Maren Schickedanz: Dünnschichtchromatographische Enantiomerentrennung mittels Ligandenaustausch. In: Angewandte Chemie. Band 96, 1984, S. 514–515, doi:10.1002/ange.19840960724.
- Kurt Günther: Thin-layer chromatographic enantiomeric resolution via ligand exchange. In: Journal of Chromatography A. Band 448, 1988, S. 11–30, doi:10.1016/S0021-9673(01)84562-3.
- Kurt Günther, Maren Schickedanz, Jürgen Martens: Thin-Layer Chromatographic Enantiomeric Resolution. In: Naturwissenschaften. Band 72, Nr. 3, 1985, S. 149–150, doi:10.1007/BF00490403.
- Teresa Kowalska, Joseph Sherma (Hrsg.): Thin Layer Chromatography in Chiral Separations and Analysis (= Chromatographic Science. Band 98). CRC Press Taylor & Francis Group, Boca Raton FL 2007, ISBN 978-0-8493-4369-8.
- H. Jork, W. Funk, W. Fischer, H. Wimmer: Dünnschicht-Chromatographie. Band 1 a: Physikalische und chemische Nachweismethoden: Grundlagen, Reagenzien. VCH Verlagsgesellschaft, Weinheim 1989, ISBN 3-527-26848-0.
- H. Jork, W. Funk, W. Fischer, H. Wimmer: Dünnschicht-Chromatographie – Reagenzien und Nachweismethoden. Band 1b, VCH Verlagsgesellschaft, Weinheim 1993, ISBN 3-527-26976-2.
- Mitchell E. Johnson: Rapid, Simple Quantitation in Thin-Layer Chromatography Using a Flatbeted Scanner. In: Journal of Chemical Education. Band 77, Nr. 3, März 2000, ISSN 0021-9584, S. 368, doi:10.1021/ed077p368.
- Paul Abu-Rabie, Neil Spooner: Direct Quantitative Bioanalysis of Drugs in Dried Blood Spot Samples Using a Thin-Layer Chromatography Mass Spectrometer Interface. In: Analytical Chemistry. Band 81, Nr. 24, 2009, S. 10275–10284. PMID 19919036, doi:10.1021/ac901985e.
- Joseph Sherma, Bernard Fried (Hrsg.): Handbook of thin-layer chromatography. (= Chromatographic Science. Band 89). 3., korr. und erweit. Auflage. CRC Press, Boca Raton FL u. a. 2003, ISBN 0-8247-0895-4, S. 323. (Eingeschränkte Vorschau in der Google-Buchsuche).