Kinetik (Chemie)
Die Kinetik ist ein Teilbereich der physikalischen Chemie, der in Makrokinetik und molekulare Kinetik unterteilt wird. Die molekulare Kinetik behandelt den zeitlichen Ablauf von chemischen Reaktionen auf der molekularen Ebene und schließt die Mikrokinetik ein, welche sich mit der Kinetik von Elementarreaktionen beschäftigt.[1] Die Makrokinetik betrachtet den Einfluss makroskopischer Wärme- und Stofftransportprozesse auf die Kinetik chemischer Reaktionen und stellt damit das Bindeglied zwischen Reaktionskinetik und chemischer Reaktionstechnik dar. Dieser Artikel beschäftigt sich mit der molekularen Kinetik chemischer Reaktionen. Die Kinetik elektrochemischer Prozesse wird im Artikel elektrochemische Kinetik behandelt.

Reaktionsgeschwindigkeit
Definition
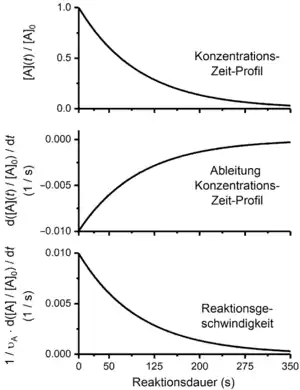






Die grundlegende Größe der Kinetik ist die Reaktionsgeschwindigkeit (englisch rate of reaction) mit der Dimension Stoffmenge pro Zeit und Volumen.[2][3] Die aus den Basisgrößen des internationalen Einheitensystems abgeleitete Einheit der Reaktionsgeschwindigkeit ist Mol pro Kubikmeter und Sekunde. Die Reaktionsgeschwindigkeit gibt die Änderung der Umsatzvariable (Menge der Reaktionsereignisse, die durch die Reaktionsgleichung der betrachteten Reaktion definiert werden, in mol) pro Zeit- und Volumeneinheit unter isochoren Bedingungen an. Sind die stöchiometrische Zahl und der Betrag der stöchiometrischen Zahl eines an der betrachteten Reaktion beteiligten Stoffes i, wird für eine Reaktion





die Reaktionsgeschwindigkeit r gleich:

Hierbei sind t die Reaktionsdauer, V das Reaktionsvolumen und [A], [B], [K] sowie [L] die volumenbezogenen Stoffmengenkonzentrationen der an der betrachteten Reaktion beteiligten Stoffe A, B, K und L. Der Differentialquotient ist gleich der Steigung des zugrundeliegenden Konzentrations-Zeit-Profils [A](t), welches [A] als Funktion von t darstellt. Da A als Ausgangsstoff verbraucht wird, sind die differentielle Konzentrationsänderung d[A] und damit der Differentialquotient negativ. Da auch die stöchiometrische Zahl eines Ausgangsstoffes konventionsgemäß ein negatives Vorzeichen hat, werden der Ausdruck und somit die Reaktionsgeschwindigkeit positiv.


Geschwindigkeitsgesetze
Die Abhängigkeit der Reaktionsgeschwindigkeit von den aktuellen Konzentrationen der Reaktanten einer Reaktion wird empirisch durch Geschwindigkeitsgesetze beschrieben. Geschwindigkeitsgesetze enthalten in der Regel eine Geschwindigkeitskonstante k oder eine Halbwertszeit t1/2, die die Kinetik des betrachteten chemischen Prozesses in charakteristischer Weise repräsentieren. Die Halbwertszeit gibt dabei den Zeitraum an, in dem die Ausgangskonzentration [A]0 eines Reaktanten A auf den halben Wert absinkt.
Phänomenologisch beobachtbare Bruttoreaktionen können komplexe Reaktionsmechanismen aufweisen, die Sequenzen aus mehreren reversiblen Elementarreaktionen umfassen. Beispiele hierfür sind Reaktionen, die dem Lindemann-Mechanismus folgen, Kettenreaktionen oder durch die Michaelis-Menten-Theorie beschreibbare enzymkatalysierte Reaktionen. Weiterhin kann die beobachtbare Reaktionsgeschwindigkeit durch Konkurrenzreaktionen beeinflusst werden. Da Geschwindigkeitsgesetze somit meist komplexe Reaktionsgeschehen abbilden, kann aus diesen nicht unmittelbar auf den Reaktionsmechanismus geschlossen werden. Häufig werden Geschwindigkeitsgesetze auf der Basis von vereinfachten Reaktionsmodellen formuliert. So kann die Kinetik von zusammengesetzten Reaktionen, die mehrere konsekutive Elementarreaktionen umfassen, von einer besonders langsam ablaufenden Elementarreaktion als geschwindigkeitsbestimmendem Schritt dominiert sein. In diesem Fall wird die Kinetik der zusammengesetzten Reaktion oft in zufriedenstellender Weise durch die einfachere Kinetik der besonders langsam ablaufenden Elementarreaktion repräsentiert. Treten im Verlauf von Reaktionen reaktive Intermediate auf, kann das Bodensteinsche Quasistationaritätsprinzip angewendet werden.
Zeitgesetze
Zeitgesetze bzw. Ratengleichungen geben die Umsatzvariable einer Reaktion oder die Konzentration eines an einer Reaktion beteiligten Stoffes als Funktion der Reaktionsdauer an. In einigen Fällen lassen sich Zeitgesetze chemischer Reaktionen aus den jeweiligen Geschwindigkeitsgesetzen durch Variablentrennung und Integration ermitteln. Umgekehrt handelt es sich bei Geschwindigkeitsgesetzen um die ersten Ableitungen der entsprechenden Zeitgesetze. Halbwertszeiten lassen sich direkt aus Zeitgesetzen ermitteln.
Temperaturabhängigkeit der Reaktionsgeschwindigkeit
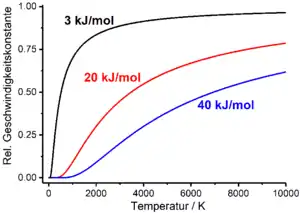
Reaktionsgeschwindigkeiten hängen grundsätzlich von den Zustandsgrößen ab, die das reagierende System kennzeichnen, wie etwa der Temperatur T, dem Druck und dem Volumen. In die Geschwindigkeitsgesetze chemischer Prozesse findet diese Abhängigkeit über die Geschwindigkeitskonstante beziehungsweise die Halbwertszeit Eingang, die ihrerseits Funktionen von Temperatur, Druck und Volumen sind. In der Praxis relevant ist vor allem die Temperaturabhängigkeit der Reaktionsgeschwindigkeit. Diese wird empirisch durch die Arrhenius-Gleichung beschrieben, die die Geschwindigkeitskonstante in Beziehung zur Temperatur setzt. Die Arrhenius-Gleichung ist eine Exponentialfunktion, welche einen präexponentiellen Faktor A mit der Dimension der Geschwindigkeitskonstante und die molare Aktivierungsenergie EA im Exponenten als empirische Parameter enthält (R ist die allgemeine Gaskonstante):

Geht man näherungsweise davon aus, dass der präexponentielle Faktor der Arrhenius-Gleichung sowie die Aktivierungsenergie unabhängig von der Temperatur sind, läuft die Geschwindigkeitskonstante gegen null, wenn die Temperatur gegen null läuft, und gegen den präexponentiellen Faktor, wenn die Temperatur gegen unendlich läuft. Der präexponentielle Faktor stellt somit den Maximalwert dar, den die Geschwindigkeitskonstante annehmen kann.
Analog kann mittels der Arrhenius-Gleichung auch dargestellt werden, wie Halbwertszeiten von der Temperatur abhängen. Die Arrhenius-Gleichung hat dann einen Exponenten mit positivem Vorzeichen:

Dabei besitzt der präexpoentielle Faktor A' wie die Halbwertszeit die Dimension Zeit. Die Halbwertszeit läuft gegen unendlich, wenn die Temperatur gegen null läuft, und gegen den präexponentiellen Faktor, wenn die Temperatur gegen unendlich läuft. Der präexponentielle Faktor stellt somit den Minimalwert dar, den die Halbwertszeit annehmen kann.
Übergangszustände und Aktivierungsenergien
Übergangszustände
Im Verlauf eines elementaren Reaktionsereignisses wird vom reagierenden System eine Trajektorie auf einer Potentialhyperfläche durchlaufen, die durch sukzessive strukturelle Änderungen wie Änderungen von Bindungswinkeln und Bindungslängen gekennzeichnet ist. Gemäß der Theorie des Übergangszustandes wird dabei ein Ausgangsstoffe und Produkte trennender Potentialwall (Aktivierungsbarriere) überwunden, der einen Sattelpunkt auf der Potentialhyperfläche darstellt. Die Zustände, die das reagierende System während der betrachteten Elementarreaktion durchläuft, werden am zweckmäßigsten durch das jeweils anzuwendende thermodynamische Potential beschrieben, welches die durch Veränderungen im reagierenden System hervorgerufenen Entropieänderungen im Universum abbildet. Werden Druck und Temperatur konstant gehalten, ist dies die freie Enthalpie. Der Sattelpunkt ist der Ort der höchsten freien Enthalpie, den das reagierende System im Verlauf eines elementaren Reaktionsereignisses durchläuft. Der Zustand, den dass reagierende System beim Durchlaufen des Sattelpunktes einnimmt, wird als Übergangszustand bezeichnet. Die molare freie Aktivierungsenthalpie repräsentiert die Höhe der für die Umwandlung der Ausgangsstoffe in die Produkte zu überwindenden Potentialbarriere, also die Differenz der freien Enthalpien des Übergangszustandes und des Ausgangszustandes vor Beginn des elementaren Reaktionsereignisses. Die Größe der Geschwindigkeitskonststante , die die Kinetik der Umwandlung der Ausgangsstoffe in die Produkte repräsentiert, hängt von ab (siehe Abschnitt „Thermodynamische Formulierung“ im Artikel „Theorie des Übergangszustandes“):



Entsprechend gilt für die Abhängigkeit der Geschwindigkeitskonststante der Rückreaktion von deren freier Aktivierungsenthalpie :



Freie Aktivierungsenthalpien und thermodynamisches Gleichgewicht
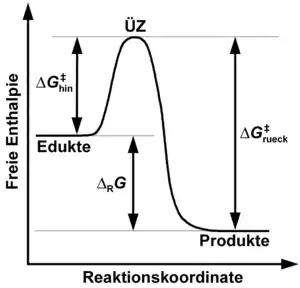

Viele Reaktionen sind Gleichgewichtsreaktionen, bei denen neben der Bildung von Reaktionsprodukten durch die Hinreaktion durch die Rückreaktion auch Ausgangsstoffe aus den Reaktionsprodukten neu gebildet werden:

Sofern die Hinreaktion mit der molaren freien Aktivierungsenthalpie sowie der molaren freien Reaktionsenthalpie


und die Rückreaktion mit der freien Aktivierungsenthalpie

exakt entlang derselben Reaktionstrajektorie in jeweils entgegensetzter Richtung ablaufen, gilt für die molare freie Aktivierungsenthalpie der Hinreaktion:

Die Geschwindigkeitskonstante der Hinreaktion wird dann:


Für den Quotienten aus khin und krueck folgt:

Vereinfachung ergibt:

Somit wird:

Dabei ist K die thermodynamische Gleichgewichtskonstante der betrachteten Reaktion. Die Geschwindigkeitskonstanten khin der Hinreaktion und krueck der Rückreaktion sind somit durch miteinander gekoppelt ‒ das Verhältnis wird durch die thermodynamische Gleichgewichtskonstante bestimmt. Dieser Zusammenhang wird oft dahingehend fehlinterpretiert, dass die Gleichgewichtskonstante einer Gleichgewichtsreaktion von den Geschwindigkeitskonstanten der Hin- und Rückreaktionen abhinge. Diese Vorstellung beruht jedoch auf einem unzutreffenden Präkonzept. Thermodynamische Größen, die wie die freie Reaktionsenthalpie und die Gleichgewichtskonstante Zustandsänderungen beschreiben, hängen ausschließlich von Ausgangs- und Endzustand ab, nicht jedoch vom Weg, auf dem sich das System vom Ausgangs- zum Endzustand bewegt.


Aktivierungsenergien
Die Existenz von Potentialbarrieren, die im Verlauf einer phänomenologisch beobachtbaren Bruttoreaktion bei der Umwandlung der Ausgangsstoffe in die Produkte überwunden werden müssen, wird empirisch durch die Arrhenius-Gleichung abgebildet. Im Gegensatz zur aus der Theorie des Übergangszustandes resultierenden Eyring-Gleichung berücksichtigt die Arrhenius-Gleichung dabei weder die Zahl noch die Natur der vom reagierenden System zu durchlaufenden Übergangszustände, sondern repräsentiert die zu überwindenden Potentialbarrieren durch den phänomenologischen Parameter Aktivierungsenergie. Läuft die Aktivierungsenergie bei konstanter Temperatur gegen unendlich, konvergiert die Geschwindigkeitskonstante gemäß der Arrhenius-Gleichung gegen null. Ist die Aktivierungsenergie gleich null, wird die Geschwindigkeitskonstante gleich dem präexponentiellen Faktor A. Analog kann mittels der Arrhenius-Gleichung auch dargestellt werden, wie Halbwertszeiten von der Aktivierungsenergie abhängen. Läuft die Aktivierungsenergie bei konstanter Temperatur gegen unendlich, läuft auch die Halbwertszeit gegen unendlich. Ist die Aktivierungsenergie null, wird die Halbwertszeit gleich dem präexponentiellen Faktor A'.
Allgemein erhöht sich bei konstanter Temperatur die Reaktionsgeschwindigkeit, wenn die Aktivierungsenergie reduziert wird. Bei der Katalyse chemischer Reaktionen werden daher alternative Reaktionstrajektorien bereitgestellt, die zu einer Verringerung der Aktivierungsenergie führen. Damit werden höhere Reaktionsgeschwindigkeiten erreicht, ohne hierfür die Reaktionstemperatur erhöhen zu müssen.
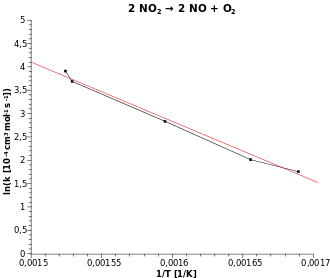
Werden für eine Reaktion bei mehreren Temperaturen Konzentrations-Zeit-Profile und aus diesen die jeweiligen Geschwindigkeitskonstanten k(T) experimentell bestimmt, lässt sich auch die Aktivierungsenergie ermitteln. Logarithmieren überführt die Arrhenius-Gleichung in eine Geradengleichung:

Die graphische Auftragung des natürlichen Logarithmus ln k(T) der experimentell ermittelten Geschwindigkeitskonstanten k(T) gegen ergibt eine Gerade, deren Steigung der negativen Aktivierungsenergie entspricht. Der Schnittpunkt der Geraden mit der y-Achse ist der Logarithmus ln A des präexponentiellen Faktors der Arrhenius-Gleichung.

Reaktionen nullter bis dritter Ordnung
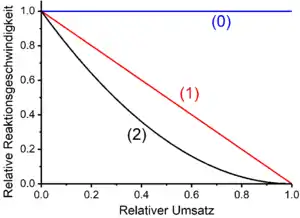

In vielen Fällen ist die Geschwindigkeit chemischer Reaktionen, an denen Ausgangsstoffe beteiligt sind, proportional zu Produkten von Potenzfunktionen der Konzentrationen Ausgangsstoffe :




Hierbei ist die Geschwindigkeitskonstante. Die Exponenten sind die partiellen Reaktionsordnungen in Bezug auf die Ausgangsstoffe . Die Summe ist die Gesamtreaktionsordnung der betrachteten Reaktion. Partielle Reaktionsordnungen können, müssen aber nicht die gleichen Beträge wie die stöchiometrischen Zahlen der betreffenden Ausgangsstoffe einer Reaktion besitzen. Die in Bruttoreaktionsgleichungen auftretenden stöchiometrischen Zahlen der Ausgangsstoffe repräsentieren häufig die Gesamtstöchiometrie zusammengesetzter Reaktionen, die mehrere Elementarreaktionen umfassen. Partielle Reaktionsordnungen und damit die Gesamtreaktionsordnungen resultieren häufig, wie bei nukleophilen Substitutionen und Eliminierungen erster Ordnung, aus geschwindigkeitsbestimmenden Elementarreaktionen, deren Stöchiometrie sich jeweils von der Stöchiometrie der betrachteten Bruttoreaktion unterscheidet. Sofern eine Bruttoreaktion wie etwa bei radikalischen Kettenreaktionen sich überlagernde oder gekoppelte Elementarreaktionen umfasst, ohne dass eine langsame und damit geschwindigkeitsbestimmernde Elementarreaktion existiert, können sich die partiellen Reaktionsordnungen ebenso von den Stöchiometriezahlen der betreffenden Komponenten unterscheiden. In derartigen Fällen treten auch gebrochene Reaktionsordnungen auf.[4][5]




Die für Reaktionen mit erster oder größerer Gesamtreaktionsordnung resultierenden Zeitgesetze lassen sich durch Umformungen in Geradengleichungen überführen (siehe Tabelle unten; für Reaktionen nullter Ordnung sind Konzentrations-Zeit-Profile immer linear). In dieser Form dargestellte Zeitgesetze können zur experimentellen Ermittlung der Gesamtreaktionsordnung mit nach verschiedenen Reaktonsdauern gemessenen Konzentrationen [A](t) eines Reaktanten A verglichen werden. Der Betrag der Steigung der so erhaltenen Geraden entspricht dem Produkt aus der Stöchiometriezahl des Ausgangsstoffes und der Geschwindigkeitskonstante .


Übersicht
Zeitgesetze, die linearisierten Auftragungen der Zeitgesetze zur Bestimmung der Geschwindigkeitskonstante sowie die Ausdrücke für die Halbwertszeichen sind für Reaktionen mit Geschwindigkeitsgesetzen des Typs in nachfolgender Tabelle unter Berücksichtigung der Stöchiometriezahl von Ausgangsstoff zusammengefasst.[6][7] Zu beachten ist, dass ein negatives Vorzeichen hat. Die Einheiten der Geschwindigkeitskonstanten sind für alle Reaktionen mit der Gesamtreaktionsordnung n gültig.

| 0. Ordnung | 1. Ordnung | 2. Ordnung | n. Ordnung | |
|---|---|---|---|---|
| Geschwindigkeitsgesetz | ||||
| Zeitgesetz |
[] | |||
| Einheit k | ||||
| Lineare Auftragung des Zeitgesetzes | [A] vs. t | vs. t | vs. t | vs. t
[] |
| Halbwertszeit |
[] |




















Reaktionen nullter Ordnung
Die Geschwindigkeit von Reaktionen nullter Ordnung ist unabhängig von den Konzentrationen der Reaktanten. Dadurch ist die Reaktionsgeschwindigkeit konstant. Zeitgesetze nehmen demzufolge die Form einer Geraden mit negativer Steigung (für Umsatzvariable und Reaktanten) oder mit positiver Steigung (für Produkte) an. Die Geradensteigung entspricht dem Betrag der Geschwindigkeitskonstante k. Beispiele für Reaktionen nullter Ordnung sind bestimmte photochemische und katalytische Reaktionen. So ist beispielsweise die biologische Oxidation von Ethanol zu Acetaldehyd durch bestimmte Alkoholdehydrogenasen nullter Ordnung in Bezug auf Ethanol.[8] Ein weiteres Beispiel ist die Emulsionspolymerisation, bei der nach der Phase der Initiierung die Reaktion 0. Ordnung ist.[9]
Reaktionen erster Ordnung
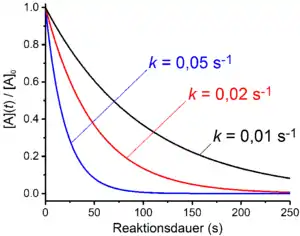
Bei Reaktionen erster Ordnung hängt die Reaktionsgeschwindigkeit linear von der Konzentration eines Reaktanden A ab. Dies ist der Fall, wenn der Gesamtprozess lediglich eine unimolekulare Zerfallsreaktion umfasst. Ein Beispiel hierfür ist der radioaktive Zerfall. Weiterhin sind Reaktionen, die mehrere Elementarreaktionen umfassen, erster Ordnung, wenn der geschwindigkeitsbestimmende Schritt ein Zerfalls- oder Dissoziationsprozess ist. Beispiele hierfür sind der SN1-Mechanismus der nukleophilen Substitution oder der E1-Mechanismus bei Eliminierungsreaktionen. Das Geschwindigkeitsgesetz nimmt für eine Reaktion erster Ordnung folgende Form an:

Das Zeitgesetz erster Ordnung wird erhalten, indem zunächst die Variablen getrennt werden:

Im nächsten Schritt berechnet man die bestimmten Integrale:

Man erhält zunächst:

Umformen ergibt:

Entlogarithmieren ergibt das Zeitgesetz für Reaktionen erster Ordnung:

Beziehungsweise:

Sofern, wie es bei Reaktionen erster Ordnung häufig der Fall ist, gilt
- ,

wird das Zeitgesetz für Reaktionen erster Ordnung:

Reaktionen zweiter Ordnung
Zweiter Ordnung sind Elementarreaktionen, die auf bimolekularen Stößen beruhen. Beispiele hierfür sind nukleophile Substitutionen nach dem SN2-Mechanismus sowie Eliminierungen nach dem E2-Mechanismus. Werden im Verlauf einer mehrschrittigen Reaktion auch Zerfalls- oder Dissoziationsprozesse erster Ordnung durchlaufen, sind diese meist die geschwindigkeitsbestimmenden Schritte, so dass, wie etwa bei SN1- und E1-Reaktionen, die Gesamtreaktion erster Ordnung ist.
Findet der bimolekulare Stoß unter Beteiligung zweier gleichartiger Teilchen der Species A statt, ist der Betrag der stöchiometrischen Zahl des Ausgangsstoffes A gleich zwei, und die Reaktionsgleichung hat die Form:


Die hieraus resultierenden Geschwindigkeits- und Zeitgesetze sowie der Ausdruck für die Halbwertszeit von [A](t) sind in der Tabelle oben aufgeführt.
Sind am bimolekularen Stoß Teilchen zweier verschiedener Species A und B beteiligt und sind die Beträge der stöchiometrischen Zahlen und der Reaktanten A und B jeweils gleich eins, wird die Reaktionsgleichung:


Das Geschwindigkeitsgesetz wird entsprechend:

Als Zeitgesetz erhält man mit [B]0 als Ausgangskonzentration des Reaktanten B vor Reaktionsbeginn:[10]

Reaktionen dritter Ordnung
Damit eine Reaktion dritter Ordnung ist, muss diese einen trimolekularen Stoß als Elementarreaktion umfassen. Da trimolekulare Stöße eine geringe Wahrscheinlichkeit besitzen, sind Reaktionen dritter Ordnung selten. Das Auftreten von Reaktionen dritter Ordnung wurde etwa im Zusammenhang mit Rekombinationsreaktionen von Atomen und Radikalen des Typs
- ,

wobei M als Stoßpartner fungiert, sowie für Reaktionen von Stickstoffmonoxid mit Halogenen X und Sauerstoff gemäß

und

postuliert.[11]
Geschwindigkeitsgesetze von Gleichgewichtsreaktionen
Betrachtet wird eine Gleichgewichtsreaktion
- .

Sind die Hin- und Rückreaktion jeweils erster Ordnung, werden die Änderungen der Konzentrationen und bestimmt durch die Differentialgleichungen




mit den Geschwindigkeitskonstanten der Reaktion und der Rückreaktion , sowie der Bedingung





Zur Lösung dieses Differentialgleichungssystems sind folgende Randbedingung nötig:
- : ,
- : ,






Die zwei Differentialgleichungen für und vereinfachen sich durch diese Randbedingung zu:



Es ergibt sich daraus das folgende integrale Zeitgesetz für die Gleichgewichtsreaktionen 1. Ordnung:

Mit
- und


sowie

ergeben sich folgende Geschwindigkeitsgesetze:


Messung von Konzentrations-Zeit-Profilen
Experimentell wird die Kinetik chemischer Reaktionen durch die Ermittlung von Konzentrations-Zeit-Profilen mittels quantitativer analytischer Methoden untersucht. Hierfür stellt die analytische Chemie ein breites und sich durch den Fortschritt der instrumentellen Analytik sowie der Mikroreaktionstechnik kontinuierlich weiterentwickelndes Methodensprektrum zur Verfügung. Die zur Ermittlung der Konzentrations-Zeit-Profile gewählte Messgröße sollte quantitativ proportional zur Konzentration der beobachteten Komponenten sein. Häufig verwendete Methoden zur Ermittlung von Konzentrations-Zeit-Profilen sind unter anderem die Messung der Dielektrizitätskonstanten, des Brechungsindexes, der optischen Aktivität, der Fluoreszenz oder der Leitfähigkeit der Reaktionslösung, die Messung von Volumen- oder Druckänderungen, Kalorimetrie sowie Absorptions- und Emissionsspektroskopie und Lichtstreuung.
Die Messgröße kann dabei am Reaktionsgemisch selbst berührungsfrei und ohne Entnahme von Proben aus dem Reaktionsgemisch beobachtetet werden. Diese Vorgehensweise ist vorteilhaft, da auf diese Weise Störungen des Reaktionsablaufs minimiert werden. Eine andere Vorgehensweise beruht darauf, dass der Reaktionsmischung regelmäßig und meist automatisiert Aliquote entnommen werden. Diese können zerstörungsfreien Analysemethoden unterzogen und danach wieder mit der Reaktionsmischung vereinigt werden. Ebenso können Analysemethoden angewendet werden, die mit der Konsumption der entnommenen Aliquote verbunden sind. Werden der Reaktionsmischung Aliquote entnommen, greift man stärker als bei berührungsfreien Verfahren in das Reaktonsgeschehen ein. Vorteilhaft ist jedoch, dass ein wesentlich breiteres Spektrum an Analysemethoden eingesetzt werden kann und Analysemethoden zur Anwendung kommen können, die entweder empflindlicher sind oder in anderer Weise mehr Informationen liefern als die für die berührungsfreie Beobachtung des Reaktionsgemisches anwendbaren Analysemethoden. So können die entnommenen Aliqote auch komplexerer qualitativer Analytik unterzogen werden, etwa indem deren Komponenten vor der eigentlichen analytischen Untersuchung mittels Gaschromatographie oder Hochleistungsflüssigkeitschromatographie aufgetrennt werden. Unter Umständen muss die Reaktion in den entnommenen Aliquoten verlangsamt oder zum Stillstand gebracht werden, um eine Verfälschung der Analyseergebnisse durch ein Fortlaufen der Reaktion nach der Probenentnahme zu unterbinden. Dies kann geschehen, indem man die entnommene Reaktionsmischung stark kühlt oder indem eine reaktive Komponente aus der Reaktionsmischung entfernt wird, beispielsweise durch Fällung.
Zur Messung von Konzentrations-Zeit-Profilen muss eine ausreichende Durchmischung der Reaktanten so schnell erfolgen, dass ein definierter Startzeitpunkt für die zu untersuchende Reaktion identifizierbar ist. Dies kann durch Miniaturisierung der verwendeten Versuchsaufbauten mittels des Einsatzes von Mikroreaktionstechnik erreicht werden, da dann die Transportwege für die Reaktanten verkürzt werden. Bei langsamen Reaktionen lassen sich definierte Stoffmengen unter Verwendung von einfachen Rührern, Strömungsrohren oder hochpräzisen Mischkammern vermischen. Bei schnelleren Reaktionen, die durch Zeitskalen im Minuten- bis Sekundenbereich charakterisiert sind, werden häufig spezielle Strömungsapparaturen verwendet. Bei extrem schnellen Reaktionen, die durch Zeitskalen im Millisekundenbereich charakterisiert sind, werden für schnelle und effiziente Mischung der Ausgangsstoffe optimierte Verfahren, wie etwa die Stopped-Flow-Methode, eingesetzt.
Eine zweite Gruppe von Verfahren zur Untersuchung extrem schneller Reaktionen mit charakteristischen Zeitskalen bis hinab zum Pikosekundenbereich sind Relaxationsverfahren. Diese beruhen auf dem Prinzip, die Ausgangsstoffe bereits deutlich vor dem eigentlichen Beobachtungszeitraum zu vermischen. Das Reaktionsgemisch beginnt zu reagieren. Nachdem sich in der Reaktionsmischung ein Gleichgewichtszustand eingestellt hat, wird dieser durch einen schnell applizierten Schock gestört und die Relaxation des Reaktionsgemisches in einen neuen Gleichgewichtszustand mit geeigneten Analysemethoden verfolgt. Beispiele für Relaxationsverfahren sind die Blitzlichtphotolyse sowie Temperatur-, Druck- und Feldsprungverfahren.
Geschichte der Kinetik
Erste qualitative Untersuchungen zur Kinetik wurden bereits um 1777 von Carl Friedrich Wenzel in seinem in Dresden erschienenem Werk Lehre von der Verwandtschaft der Körper berichtet. Später beschäftigten sich auch Claude-Louis Berthollet und William Higgins mit kinetischen Fragestellungen.[12] Die erste wirklich grundlegende Arbeit zur Kinetik, die Spaltung von Rohrzucker unter Säureeinfluss, wurde von Ludwig Ferdinand Wilhelmy im Jahr 1850 vorgelegt.[13] Jacobus Henricus van't Hoff untersuchte im Jahr 1896 die Verseifung von Essigester und die Hydrolyse von Chloressigsäure. Mathematisch formulierte er die Geschwindigkeitsgleichungen der Reaktionen. Ferner entwickelte er die grundlegenden Gesetze zu Temperaturabhängigkeit der Reaktionsgeschwindigkeit.[14] Svante Arrhenius verbesserte die Ableitung und gab als Faustformel für die Änderung der Reaktionsgeschwindigkeit bei Temperaturerhöhung um 1 K eine Erhöhung der Reaktionsgeschwindigkeit um ca. 12 % an (siehe RGT-Regel). F. E. C. Scheffer und W. F. Brandsma führten im Jahr 1926 die Standard-Gibbs-Aktivierungsenergie für die Geschwindigkeitskonstante ein.[15] Für die Entwicklung der Relaxationsverfahren zur Untersuchung der Kinetik schneller Reaktionen erhielten Manfred Eigen, Ronald Norrish und George Porter 1967 den Nobelpreis für Chemie.
Literatur
Allgemeine Lehrbücher
Kinetik
- Margaret Robson Wright: An Introduction to Chemical Kinetics. John Wiley & Sons, Ltd, Chichester, UK 2004, ISBN 978-0-470-09060-2, doi:10.1002/047009060x.ch1.
- Bernd Ralle, Ingo Eilks, Alfred Flint, Hartwig Möllencamp, Helmut Wenck: Handbuch der experimentellen Chemie: Sekundarbereich II. 8, Kinetik, Katalyse, Gleichgewicht. Hrsg.: Bernd Ralle, Ingo Eilks, Wolfgang Glöckner, Walter Jansen, Rudolf G. Weissenhorn. Aulis-Verlag Deubner, Köln 2004, ISBN 3-7614-2384-5.
- Carl Heinz Hamann, Dirk Hoogestraat, Rainer Koch: Grundlagen der Kinetik: Von Transportprozessen zur Reaktionskinetik. Springer-Verlag, Berlin 2017, ISBN 978-3-662-49393-9, doi:10.1007/978-3-662-49393-9.
- M. Dieter Lechner: Einführung in die Kinetik: Chemische Reaktionskinetik und Transporteigenschaften. Springer-Verlag, Berlin/Heidelberg 2018, ISBN 978-3-662-57454-6, doi:10.1007/978-3-662-57455-3_1.
Weblinks
- G. J. Lauth: Einführung in die Reaktionskinetik. Vorlesungsreihe, Videoaufzeichnungen, 2013.
Einzelnachweise
- K. J. Laidler: A glossary of terms used in chemical kinetics, including reaction dynamics (IUPAC Recommendations 1996). In: Pure and Applied Chemistry. Band 68, Nr. 1, 1. Januar 1996, ISSN 1365-3075, S. 149–192, doi:10.1351/pac199668010149.
- Eintrag zu rate of reaction. In: IUPAC (Hrsg.): Compendium of Chemical Terminology. The “Gold Book”. doi:10.1351/goldbook.R05156 – Version: 2.3.3.
- DIN 13345-1978-08 Thermodynamik und Kinetik chemischer Reaktionen: Formelzeichen, Einheiten.
- Carl Heinz Hamann, Dirk Hoogestraat, Rainer Koch: Grundlagen der Kinetik: Von Transportprozessen zur Reaktionskinetik. Springer-Verlkag, Berlin/Heidelberg 2017, ISBN 978-3-662-49392-2, S. 116, doi:10.1007/978-3-662-49393-9.
- M. Dieter Lechner: Einführung in die Kinetik - Chemische Reaktionskinetik und Transporteigenschaften. Springer-Verlag, Berlin/Heidelberg 2018, ISBN 978-3-662-57454-6, S. 13, doi:10.1007/978-3-662-57455-3_1.
- Christos Capellos, Benon H. J. Bielski: Kinetic systems: mathematical description of chemical kinetics in solution. Wiley-Interscience, 1972, ISBN 0-471-13450-3, 9780471134503.
- Robert G. Mortimer: Physical Chemistry. Academic Press, 2000, 2. Auflage, Kapitel 12 The Rates of Chemical Reactions, S. 408.
- Ignacio Tinoco Jr., James C. Wang: Physical chemistry: principles and applications in biological sciences. Prentice Hall, 1995, 3. Auflage, S. 331, ISBN 978-0-13-186545-7.
- Alisa Gapchenko Styrol-Butylacrylat-Emulsion: Aspekte der Hochtemperatur-Copolymerisation und neue Anwendungsgebiete Dissertation des Fachbereich Chemie der Fakultät für Mathematik, Informatik und Naturwissenschaften der Universität Hamburg 2018, abgerufen am 29. August 2020
- Lothar Papula: Übungen und Anwendung zur Mathematik für Chemiker. Enke Verlag, 1977, S. 382–385.
- Klaus H.Homann: 13. Trimolekulare Reaktionen. S. 100–107, in Reaktionskinetik, Steinkopff-Verlag, Heidelberg 1975, DOI 10.1007/978-3-642-72314-8_13, https://doi.org/10.1007/978-3-642-72314-8_13.
- V. A. Kritsman: Ludwig Wilhelmy, Jacobus Henricus van't Hoff, Svante Arrhenius und die Geschichte der chemischen Kinetik. In: Chemie in unserer Zeit. Bd. 6, 1997, S. 291 ff.
- L. Wilhelmy: Über das Gesetz, nach welchem die Einwirkung der Säuren auf den Rohrzucker stattfindet. In: Pogg. Ann. 81, 1850, S. 413–433, 499–526.
- J. H. van't Hoff: Studien zur chemischen Dynamik. W. Engelmann, Leipzig 1896.
- F. E. C. Scheffer, W. F. Brandsma: On reaction velocities. In: Recueil des Travaux Chimiques des Pays-Bas 1926, 45, 522–534, DOI:10.1002/recl.19260450710.