D-Aminosäuren
D-Aminosäuren sind eine Klasse von Aminosäuren, bei denen die funktionellen Gruppen – Carboxygruppe (–COOH) und Aminogruppe (–NH2) – α-ständig in D-Konfiguration vorliegen. Es sind Spiegelbildisomere der L-Aminosäuren.
D-Aminosäuren sind in allen bekannten biologischen Systemen wesentlich seltener als ihre L-Isomere vertreten, die in Form der 23 proteinogenen Aminosäuren wichtige Bausteine des Lebens sind. Man ging deshalb lange Zeit davon aus, dass D-Aminosäuren überhaupt keine biologische Funktion haben und „unnatürlich“ sind. Seit dem Beginn der 1990er Jahre hat sich dieses Bild gewandelt. Heute weiß man, dass D-Aminosäuren zum Beispiel in von Bakterien hergestellten Peptid-Antibiotika enthalten sind sowie in verschiedenen Pflanzen, wie Reis, Knoblauch und Erbsen.
Einige D-Aminosäuren erfüllen auch beim Menschen wichtige physiologische Funktionen. Insbesondere im Zentralnervensystem sind dies D-Serin und D-Asparaginsäure. D-Aminosäuren scheinen aber auch bei bestimmten Erkrankungen, wie zum Beispiel Schizophrenie, eine Rolle zu spielen. Dieses Forschungsgebiet ist vergleichsweise neu, und viele Funktionen der freien und in Peptiden oder Proteinen gebundenen D-Aminosäuren sind noch weitgehend unbekannt oder unverstanden.
Mit Hilfe chromatografischer Analyseverfahren konnten D-Aminosäuren in einer Reihe von Lebensmitteln und Organismen nachgewiesen werden. Eine Anwendung hierbei ist die Aminosäuredatierung zur Bestimmung des Alters von Fossilien.
Freie D-Aminosäuren sind nach dem heutigen Stand der Wissenschaft in den täglich mit der Nahrung aufgenommenen Mengen für den Menschen ungefährlich. Technisch produzierte D-Aminosäuren werden als Bausteine zur Herstellung (halb)synthetischer Antibiotika verwendet und sind chemisch gebundener Bestandteil einer Vielzahl anderer Arzneistoffe.
Grundlagen
Chiralität
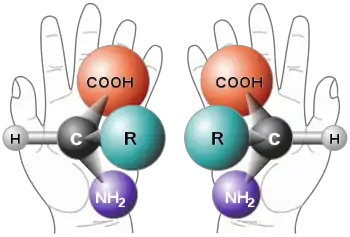
Alle proteinogenen Aminosäuren haben, mit Ausnahme von Glycin, der einfachsten Aminosäure, mindestens ein Kohlenstoffatom, das vier unterschiedliche Atome oder Atomgruppen (Substituenten) trägt. Diese Substituenten nehmen räumlich betrachtet die vier Ecken eines Tetraeders ein. Diese Anordnung bewirkt eine Asymmetrie, die zwei unterschiedliche Möglichkeiten der Ausrichtung der Substituenten zur Folge hat. Diese beiden Formen, Enantiomere oder auch Spiegelbildisomere genannt, verhalten sich wie Bild und Spiegelbild. Das asymmetrische Kohlenstoffatom bildet dabei das sogenannte Stereozentrum. Bild und Spiegelbild der Enantiomere lassen sich nicht zur Deckung bringen. Dies ist auch bei Gegenständen des täglichen Lebens der Fall, die keine Drehspiegelachse aufweisen. Ein Beispiel dafür sind die Hände. Die linke und die rechte Hand sind wie Bild und Spiegelbild, sie lassen sich jedoch nicht zur Deckung bringen. Besonders deutlich werden die Unterschiede von rechter und linker Hand, wenn sie mit anderen chiralen (das griechische Wort für ‚händig‘) Systemen interagieren. So beispielsweise, wenn eine rechte Hand eine zweite rechte oder linke Hand schüttelt oder versucht einen „falschen“ Handschuh anzuziehen. In chiralen Umgebungen werden auch bei den molekularen Enantiomeren Unterschiede deutlich.
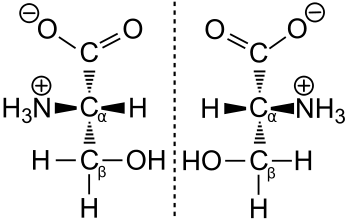
Der deutsche Nobelpreisträger für Chemie Emil Fischer entwickelte ein Projektionsverfahren, die Fischer-Projektion, mit der man die Raumstruktur einer chiralen chemischen Verbindung eindeutig zweidimensional abbilden kann. Dabei wählte er eine Bezugssubstanz (Glyceraldehyd) aus. Gemäß den Regeln bei der Fischer-Projektion wird die Säuregruppe (Carboxygruppe) immer oben gezeichnet und der die Aminosäuren unterscheidende Rest R immer unten. Liegt die Aminogruppe bei diesem Projektionsverfahren links (lat. laevus), so spricht man von einer L-Aminosäure. Der Buchstabe L wird dabei in Kapitälchen dem Namen der Aminosäure vorangestellt; beispielsweise L-Serin. Liegt die Aminogruppe bei der Fischer-Projektion auf der rechten Seite (lat. dexter = ‚rechts‘), so handelt es sich um eine D-Aminosäure. Die Adjektive links und rechts beziehen sich dabei einzig auf die nach der Fischer-Projektion dargestellte Konfiguration.
In ihren physikalischen Eigenschaften, wie beispielsweise Schmelzpunkt, Dichte, Löslichkeit in Wasser und anderen Lösungsmitteln sowie isoelektrischer Punkt, sind D- und L-Aminosäuren völlig identisch. Auch in einer achiralen Umgebung, das heißt in einem Umfeld, in dem keine anderen chiralen Moleküle vorhanden sind, verhalten sie sich mit einer Ausnahme gleich: Die beiden Enantiomere drehen die Polarisationsebene von linear polarisiertem Licht unter gleichen Bedingungen (Konzentration, Temperatur, pH-Wert, Lösungsmittel usw.) dem Beitrag nach gleich, aber in unterschiedliche Richtungen. Drehen sie das Licht im Uhrzeigersinn, so spricht man von rechtsdrehend oder der (+)-Form. Die gegen den Uhrzeigersinn drehende Form nennt man linksdrehend oder die (−)-Form. Drehsinn und Drehrichtung von Aminosäuren spielen in der täglichen Praxis kaum eine Rolle. Wesentlich wichtiger ist die Konfiguration – D- oder L. Die Drehrichtung einer Aminosäure (links- oder rechtsdrehend) ist völlig unabhängig von der Konfiguration der Aminosäure. In der Literatur wird dies sehr häufig falsch wiedergegeben. Oft wird von „linksdrehenden Aminosäuren“ gesprochen, wenn L-Aminosäuren gemeint sind. Tatsächlich sind Drehsinn und Drehrichtung stark abhängig von der äußeren Umgebung. So weist die Aminosäure L-Leucin bei Raumtemperatur in sechsmolarer Salzsäure einen spezifischen Drehwinkel von +15,1° (= linksdrehend) und in neutralem Wasser einen von −10,8° (= rechtsdrehend) auf. In 3-molarer Natronlauge ist sie dagegen mit 7,6° wiederum linksdrehend.[1]
Ein Gemisch aus 50 % D- und 50 % L-Aminosäuren bezeichnet man als Racemat. Racemate entstehen unter anderem bei konventionellen technischen Synthesen von Aminosäuren. Sie sind optisch inaktiv, das heißt, sie sind nicht in der Lage, die Schwingungsebene polarisierten Lichtes zu drehen. Racemate haben im Vergleich zu den reinen Enantiomeren teilweise verschiedene physikalische Eigenschaften (Beispiel: Schmelzpunkt), jedoch durchgängig unterschiedliche physiologische Eigenschaften.
Namenskonventionen und Nomenklatur
Die Fischer-Projektion ist bis heute bei Aminosäuren und Sacchariden das bevorzugte Projektionssystem. Daneben wird auch die Cahn-Ingold-Prelog-Konvention (CIP-System) für Aminosäuren angewendet, die die absolute Konfiguration chiraler Moleküle beschreibt. Nach dem CIP-System sind die meisten proteinogenen L-Aminosäuren (S)-Aminosäuren. Ihre Spiegelbilder, die D-Aminosäuren weisen fast durchgängig eine (R)-Konfiguration auf. Ausnahmen sind L-Cystein, L-Cystin und L-Selenocystein, da Schwefel beziehungsweise Selen nach der CIP-Nomenklatur eine höhere Priorität als Sauerstoff haben. Diese drei L-Aminosäuren liegen in der (R)-Konfiguration vor. Die drei entsprechenden D-Aminosäuren haben dagegen die (S)-Konfiguration.
In Aminosäuresequenzen erhalten die D-Aminosäuren im Dreibuchstabencode ein vorangestelltes kleingeschriebenes großes D (Kapitälchen).
Am Beispiel des Heptapeptids Dermorphin
H-Tyr-D-Ala-Phe-Gly-Tyr-Pro-Ser-NH2
Im Einbuchstabencode werden D-Aminosäuren mit dem Kleinbuchstaben der L-Aminosäuren versehen.
Im Beispiel Dermorphin:
YaFGYPS-NH2
Natürliches Vorkommen und Entdeckungsgeschichte
D-Aminosäuren sind in der Natur weitaus seltener als die isomeren L-Aminosäuren, bei denen die proteinogenen Aminosäuren – zusammen mit den Nukleinsäuren – die Grundbausteine des Lebens darstellen. Eine ähnliche Asymmetrie bei dem Vorkommen zweier Typen von Enantiomeren gibt es bei den Kohlenhydraten. Hier ist die D-Form, beispielsweise die D-Glucose, die „natürliche“ Konfiguration. Es wird geschätzt, dass D-Glucose auf der Erde um den Faktor 1015 häufiger als L-Glucose ist.[2] Für Aminosäuren gibt es hier noch keine zuverlässigen Schätzungen.
Lange Zeit ging man davon aus, dass nur die L-Aminosäuren während der Evolution für die Bildung von Peptiden und Proteinen ausgewählt wurden.[3] Verbesserte Analysenmethoden führten seit den 1980er Jahren zu einer Revidierung dieser Annahme. In immer mehr Lebewesen wurden D-Aminosäuren nachgewiesen, so dass sie eine deutlich größere Verbreitung und Häufigkeit haben als ursprünglich angenommen. In der neueren Literatur werden D-Aminosäuren deshalb als gewöhnlicher Bestandteil von Pflanzen und Nahrungsmitteln angesehen.[2] Doch auch in höheren Lebewesen, bis hin zum Menschen, sind D-Aminosäuren in wichtige physiologische Vorgänge involviert, die zum Teil noch weitgehend unverstanden sind.[4]
Die Entwicklung des Lebens auf der Erde setzte eine Homochiralität, das heißt eine einheitliche Konfiguration von Aminosäuren und anderen Bausteinen des Lebens, voraus. In einem racemischen Umfeld kann keine Selbstreplikation stattfinden.[2][5][6] Über die primäre Ursache des extremen Ungleichgewichtes der Häufigkeit der beiden isomeren Formen der Aminosäuren gibt es eine Reihe von Hypothesen. Weitgehende Einigkeit herrscht ab dem Punkt, an dem es in der Natur ein erstes kleines Ungleichgewicht zwischen D- und L-Konfiguration gab. Ab hier lässt sich durch die chirale Amplifikation – quasi ein selbstverstärkender Effekt, der in einer chemischen Reaktion zu einer weiteren Zunahme der Enantiomerenform führt, die zuvor in leichtem Überschuss vorlag – sehr gut die extreme Anreicherung einer Enantiomerenform erklären. Völlig unklar ist indes, wie es zum Bruch der Spiegelsymmetrie kam, der, mit großer Wahrscheinlichkeit weit vor dem Beginn des Lebens auf der Erde,[7] zu einem ersten leichten Überschuss der L-Konfiguration bei den Aminosäuren führte. Als mögliche Gründe für den Bruch der Spiegelsymmetrie werden unter anderem die Paritätsverletzung beim β-Zerfall (Vester-Ulbricht-Hypothese)[8][9] und das „Animpfen der Ursuppe“ mit extraterrestrischen L-Aminosäure-Überschüssen, diskutiert. Für die letztgenannte Theorie spricht, dass beispielsweise im Murchison-Meteoriten ein Überschuss des jeweiligen L-Enantiomers der nicht-proteinogenen Aminosäuren 2-Amino-2,3-dimethylpentansäure[10] und Isovalin[11] nachgewiesen werden konnte.[12] Im Murchison-Meteoriten betrug der Überschuss an L-Isovalin etwa 18,5 und im Orgueil-Meteoriten ca. 15,2 Prozent.[13] Dieser Überschuss wurde möglicherweise durch zirkularpolarisierte UV-Strahlung erzeugt, die – experimentell bestätigt – bevorzugt D-Aminosäuren zerstört.[14]
Bildung von D-Aminosäuren durch Racemisierung
Größere Mengen an D-Aminosäuren können durch Racemisierung aus L-Aminosäuren entstehen. Die Bildung eines Aminosäureracemates, also eines Gemisches, das 50 % D- und 50 % L-Aminosäure enthält, ist thermodynamisch bevorzugt. Die Enthalpie bleibt zwar unverändert, aber der höhere „Grad an Unordnung“ führt zu einem Anstieg der Entropie, wodurch die Freie Enthalpie ΔG des Systems abnimmt. Der Wert beträgt bei 25 °C etwa −1,6 kJ/mol.[15] Höhere Temperaturen führen zu einer höheren Abgabe an Freier Enthalpie, weshalb die Racemisierung deutlich beschleunigt abläuft. Die Halbwertszeit der Racemisierung, die als die Zeit definiert ist, in der der ee-Wert von 100 auf 50 % sinkt, ist neben der Temperatur vor allem vom pH-Wert, der Aminosäure, dem Lösungsmittel beziehungsweise der Feuchtigkeit und der Anwesenheit von Katalysatoren abhängig. Unter konstanten Bedingungen lässt sich die Racemisierung gut vorausberechnen, beziehungsweise kann umgekehrt aus dem Grad der Racemisierung auf das Alter der untersuchten Probe geschlossen werden. Dieses als Aminosäuredatierung bezeichnete Verfahren kann zur Altersbestimmung von fossilen Proben, aber auch am lebenden Organismus herangezogen werden. Mit dem Tod enden alle Prozesse, die gegen die Racemisierung der Aminosäuren in dem betroffenen Organismen wirken. Das Leben ist ein Kampf gegen die Entropie,[16] und spätestens mit dem Tod enden die Prozesse, die einer Racemisierung entgegenwirken. In einigen Geweben mit äußerst geringem Proteinstoffwechsel beginnt dieser Prozess bereits nach Abschluss des Gewebeaufbaus. Ein Beispiel hierfür ist das Kollagen im Dentin der Zähne oder die Linse des Auges.[17] Die relativ konstanten Temperatur- und pH-Werte in den Zähnen ermöglichen auch am lebenden Organismus über den Racemisierungsgrad von Asparaginsäure eine Altersbestimmung mit einer Genauigkeit von etwa ±4 Jahren.[18][19] Das Verfahren kommt unter anderem in der Forensik zum Einsatz.[20] Ein Beispiel für die Leistungsfähigkeit dieser Methode sind Untersuchungen, die 1996 an den Gebeinen von Kaiser Lothar von Supplinburg (1075–1137) durchgeführt wurden.[21] Dabei wurde bei Lothar, im Vergleich zu seiner Frau Richenza und Heinrich dem Stolzen, ein wesentlich höherer Racemisierungsgrad festgestellt, der einem Alter von etwa 9000 Jahren entsprechen würde. Der Racemisierungsgrad der beiden Vergleichsproben entsprach dagegen sehr gut deren Alter von ca. 850 Jahren. Gemessen wurde in allen drei Fällen der Racemisierungsgrad der L-Asparaginsäure. Der hohe Racemisierungsgrad bei Lothar ist in den besonderen Umständen seines Todes begründet. Er verstarb bei Breitenwang in Tirol, etwa 700 km von seinem Stammsitz in Königslutter am Elm entfernt. Um seinen Leichnam vor dem langen Transport vor Verwesung zu schützen, wurde die Leiche nach „deutscher Sitte“ (mos teutonicus) behandelt. Dabei wurde die Leiche Lothars gekocht, das Fleisch von den Knochen entfernt und die Gebeine nach Königslutter überführt. Durch das Kochen racemisierte die 859 Jahre später gemessene L-Asparaginsäure wesentlich stärker als bei den normal bestatteten Leichen von Frau und Schwiegersohn. Über den Racemisierungsgrad konnte die Kochdauer zu etwa sechs Stunden bestimmt werden.[2]
In den Haaren der ca. 5300 Jahre alten Leiche des Mannes vom Tisenjoch, besser bekannt als „Ötzi“, liegen 37 % des Hydroxyprolins in der D-Konfiguration vor. Bei einer 3000 Jahre alten Mumie wurden 31 %, in Haaren aus dem Mittelalter (ca. 1000 Jahre alt) 19 % und in frischen Haarproben 4 % gemessen.[22]
Auch bei der Zubereitung von Lebensmitteln können unter dem Einfluss von Temperatur und extremen pH-Werten die L-Aminosäuren in den Proteinen racemisieren. Die einzelnen Aminosäuren racemisieren dabei unterschiedlich schnell. Die Racemisierungsgeschwindigkeit ist stark abhängig von der Seitenkette der jeweiligen Aminosäure und den Aminosäuren in ihrer Nachbarschaft. Elektronenziehende Gruppen vereinfachen die Protonierung des α-C-Atoms, wodurch die Racemisierung erleichtert wird.[23][24] Dies trifft beispielsweise für Serin und Asparaginsäure zu. Daneben spielen auch sterische Effekte eine Rolle.[25] Besonders leicht racemisieren Asparagin und Asparaginsäure, wenn in unmittelbarer Nachbarschaft ein Glycin in der Peptidsequenz enthalten ist. Dann kann es zur Bildung eines zyklischen Succinimids kommen, das die Epimerisierung thermodynamisch stark begünstigt.[26][27] Bei niedrigen pH-Werten, beispielsweise in sechsmolarer Salzsäure, racemisiert Asparaginsäure am stärksten. Deutlich langsamer racemisieren Prolin und Glutaminsäure, während bei diesen Bedingungen Isoleucin, Valin, Serin und Threonin nur sehr wenig racemisieren. In einmolarer Natronlauge racemisiert dagegen Serin am schnellsten, dann folgen Asparaginsäure, Phenylalanin, Glutaminsäure und Valin.[27][28]
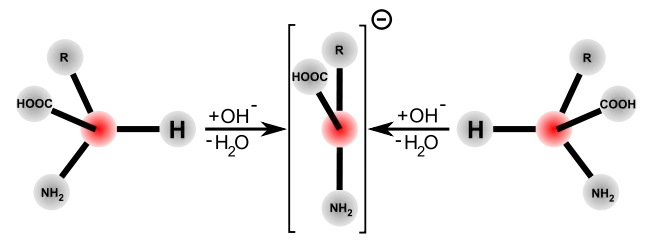
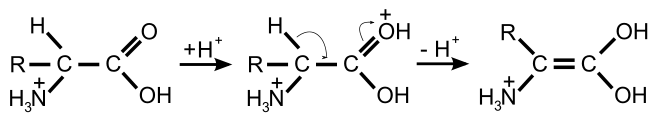
Die basen- und die säurenkatalysierte Racemisierung benötigen recht drastische Reaktionsbedingungen, um in wenigen Stunden eine vollständige Racemisierung zu erhalten. Im Gegensatz dazu verläuft die enzymkatalysierte Racemisierung in biologischen Systemen wesentlich schneller und unter sehr milden Bedingungen – im neutralen pH-Bereich und bei Raum- beziehungsweise Körpertemperatur. Die Racemasen katalysieren die Deprotonierung am α-C-Atom der Aminosäure. Das Wasserstoffatom ist in dieser Position nur äußerst schwach sauer. Die Säurekonstante der protonierten Form hat einen pKs-Wert von ≈21 und ist am isoelektrischen Punkt mit ≈29 noch schwächer.[30][31] Die Abspaltung des Protons wird bei den meisten Racemasen durch Pyridoxalphosphat (PLP) wesentlich erleichtert. Im aktiven Zentrum dieser Enzyme ist das PLP an einen Lysinrest gebunden. Die Aminogruppe der L-Aminosäure bindet dabei an die Aldehyd-Gruppe des PLP und bildet so eine Schiffsche Base (Aldimin). Als elektrophiler Katalysator zieht das PLP über den aromatischen Ring Elektronen vom α-C-Atom der Aminosäure ab, das dadurch wesentlich leichter deprotoniert. Zudem wird das verbleibende Anion mesomer stabilisiert. Eine Reprotonierung und Addition von Wasser setzt dann die racemisierte Aminosäure als Reaktionsprodukt durch Hydrolyse der Schiffschen Base frei.[32]
Daneben gibt es noch PLP-unabhängige Racemasen, in deren aktivem Zentrum die Thiolgruppen zweier Cysteine die Protonierung katalysieren.[33][34] In einem Zwei-Basen-Mechanismus nimmt dabei zunächst ein deprotoniertes Thiolat (R-S−) als Base das Proton des α-C-Atoms auf. Die Thiolgruppe des zweiten Cysteins ist dann für die Reprotonierung zuständig.[32]
Diese enzymkatalysierten Racemisierungsprozesse erzeugen den weitaus größten Teil an D-Aminosäuren in Organismen.

Peptid-Antibiotika und andere peptidische Arzneimittel natürlichen Ursprungs
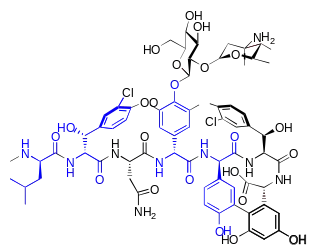
Eine große Anzahl von Peptid-Antibiotika ist aus D-Aminosäuren aufgebaut. Peptid-Antibiotika sind Naturstoffe, die von Prokaryoten mittels nichtribosomaler Peptidsynthese erzeugt werden.[32] Die pharmakologisch sehr wichtige Gruppe der Penicilline enthält als elementaren Baustein D-Penicillamin, eine nicht-proteinogene α-Aminosäure. Die Polymyxine (bei Polymyxin B1 D-Phenylalanin) und Actinomycine (D-Valin) sind ebenfalls aus D-Aminosäuren aufgebaut. Das von Bakterien der Art Bacillus subtilis gebildete Bacitracin besteht unter anderem aus D-Asparaginsäure, -Glutaminsäure, -Ornithin und -Phenylalanin. Das von Streptomyces fulvissimus produzierte Valinomycin enthält D-Valin und das von Bacillus circulans gebildete Circulin A (D-Leucin). Auch Fungisporin (D-Phenylalanin und D-Valin), Gramicidin und Tyrocidin (beide D-Phenylalanin), Malformin C (D-Leucin und D-Cystein), Mycobacillin (D-Asparaginsäure und D-Glutaminsäure) sind Peptid-Antibiotika mit D-Aminosäuren.[35]
Das von Schlauchpilzen, wie beispielsweise Tolypocladium inflatum, ausgeschiedene Immunsuppressivum Ciclosporin enthält D-Alanin. Im Isopenicillin N ist D-Valin enthalten.
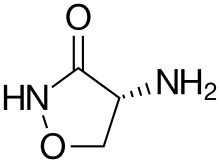
Das zur Behandlung von Tuberkulose verwendete und chemisch relativ einfach aufgebaute Cycloserin wird von Streptomyceten, wie beispielsweise Streptomyces garyphalus, aus D-Serin produziert,
D-Aminosäuren und D-Aminosäuren-haltige Peptide

Lange Zeit ging man davon aus, dass in der Natur nur ein Aminosäurenenantiomer, nämlich die L-Form, vorherrscht. D-Aminosäuren wurden bis in die 1960er Jahre hinein als „Laborartefakt“ (systemabhängiger Fehler) betrachtet und als „unnatürliches Isomer“ eingestuft.[36] Auch heute noch findet man die Bezeichnung „unnatürliche Aminosäuren“ für die D-Aminosäuren.[37]
D-Aminosäureoxidasen – Enzyme ohne Substrat?
1933 wurde von dem deutschen Mediziner, Chemiker und späteren Nobelpreisträger für Physiologie oder Medizin Hans Adolf Krebs das Enzym D-Aminosäureoxidase entdeckt[38] und zwei Jahre später ausführlich beschrieben[39].[40] Krebs stellt fest, dass die „nicht in der Natur vorkommenden“ D-Aminosäuren, in Anwesenheit von pürierter frischer Schweineniere oder -leber, deutlich schneller als ihre „natürlichen“ L-Isomere desaminiert werden. Durch gezielte Inhibierung, beispielsweise mit 1-Octanol, konnte er die in dem Püree enthaltene L-Aminosäurenoxidase deaktivieren und so erreichen, dass selektiv nur noch die D-Aminosäuren desaminiert wurden. Daraus schloss Krebs, dass sich in den verwendeten Organen, beziehungsweise deren Extrakten, zwei Aminosäureoxidasen befanden, die L- und die D-Aminosäureoxidasen, die jeweils selektiv L- bzw. D-Aminosäuren als Substrat haben. Krebs zeigte sich verwundert darüber, dass es ein Enzym gibt, das ausschließlich „nicht natürliche Substanzen“ als Substrat hat. Er verwies aber darauf, dass Felix Ehrlich 1914,[41] Edmund Oskar von Lippmann 1884[42] und Sigmund Fraenkel 1923/24[43] das gelegentliche Vorkommen von D-Aminosäuren in der Natur beschrieben.[39] Auch das von E. Winterstein und Kollegen 1913 aus Steinpilzen (Boletus edulis) isolierte D-Alanin[44], war einer dieser frühen Nachweise.[45]
D-Aminosäuren in Pflanzen
In Pflanzen lassen sich D-Aminosäuren sowohl in freier Form, als auch peptidgebunden nachweisen.[46] Häufig sind sie in Form von N-Malonyl- oder N-Acetyl-Derivaten in den Pflanzen enthalten. Beispielsweise liegt in der Wurzel der Sonnenblume (Helianthus annuus) 40 % des Alanins in der D-Konfiguration vor. D-Alanin und das Dipeptid D-Ala-D-Ala finden sich in verschiedenen Gräsern;[47] so auch in Reis (Oryza australiensis). Im Reis liegen ca. 10 % des Serins als D-Enantiomer vor. Es wird mit Hilfe einer Serin-Racemase von der Pflanze selbst erzeugt. Das entsprechende Gen für dieses Enzym liegt bei Oryza sativa ssp. Japonica cv. Nipponbare auf Chromosom 4.[48] In geringeren Konzentrationen wurden D-Aminosäuren in einer Vielzahl von Pflanzen nachgewiesen, die als Nahrungsmittel genutzt werden. Dazu gehören beispielsweise Erbsen (Pisum sativum),[49] Knoblauch, verschiedene Kohlarten und Obst.[50] Welche Funktion die freien und peptidischen D-Aminosäuren in Pflanzen haben, ist noch weitgehend unklar.[27]
Bakterien und D-Aminosäuren
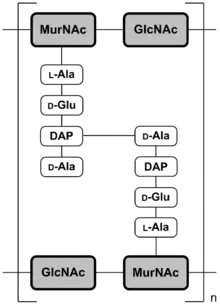
Vor dem Nachweis der freien D-Aminosäuren wurden in einer Reihe von Verbindungen mikrobiellen Ursprungs D-Aminosäuren identifiziert. Beispielsweise enthält das in Schimmelpilzkulturen gebildete, und 1928 von Alexander Fleming als erstes Penicillin entdeckte Benzylpenicillin, als wesentliches Element D-Penicillamin (= 3-Mercapto-D-Valin). Der Biochemiker Esmond E. Snell bemerkte 1943 bei Versuchen mit Kulturen von Streptococcus faecalis und Lactobacillus casei, dass das für das Wachstum dieser Bakterienstämme notwendige Pyridoxin (Vitamin B6) vollständig durch D-Alanin als Nährstoff ersetzt werden kann.[51][52][36] Er stellte außerdem fest, dass D-Alanin dabei deutlich potenter als L-Alanin war.[53] Als man danach in Peptidoglycanen – das sind die Biopolymere, die der Zellwand von Bakterien ihre Festigkeit verleiht – große Anteile von D-Alanin nachweisen konnte, war klar, wofür die Zellen diese „unnatürliche“ Aminosäure benötigen. Der Einbau von D-Alanin, und vor allem auch von D-Glutamat, verhindert den enzymatischen Abbau der Peptidoglycane durch Peptidasen. Interessanterweise ist genau dieser „Schutzwall“ aus D-Aminosäuren der Angriffspunkt für β-Lactam-Antibiotika, wie beispielsweise Penicillin. Diese Antibiotika inhibieren das Enzym D-Alanin-Transpeptidase, das ausschließlich in Bakterien vorhanden ist und die Quervernetzung der Peptidoglycane, speziell über das D-Alanin, katalysiert.[54] 1951 isolierten Irwin Clyde Gunsalus und Willis A. Wood aus Streptococcus faecalis Alaninracemase, ein Enzym, das die Racemisierung des natürlichen L-Alanins in das isomere D-Alanin katalysiert.[55] Das alr-Gen, das für die Alaninracemase codiert, ist in allen Bakterien vorhanden.[56] Das mit Hilfe der Alaninracemase gebildete D-Alanin ist für die Synthese der Peptidoglycane nahezu aller Bakterien essentiell.[57] Außer D-Alanin und D-Glutaminsäure findet sich bei bestimmten Stämmen von Enterokokken noch D-Serin in der Zellwand.[58][59] Das D-Serin bildet dabei mit D-Alanin am C-Terminus ein D-Ala-D-Ser-Dipeptid, das für die Resistenz dieser Bakterienstämme gegenüber Glykopeptid-Antibiotika, wie beispielsweise Vancomycin, verantwortlich ist.[60][61]
D-Aminosäuren in Schwämmen
In Schwämmen konnten so genannte Polytheonamide nachgewiesen werden. Das sind Peptid-Toxine, deren Aminosäuren zwischen D- und L-Form abwechseln. Sie werden offenbar ribosomal als L-Peptide synthetisiert und dann posttranslational jede zweite Aminosäure epimerisiert. Dies geschieht mithilfe weniger Enzyme, deren Gene, offensichtlich von Bakterien abstammend, über horizontalen Gentransfer in die Schwämme gelangt sind.[62][63]
D-Aminosäuren in Vielzellern
Dankwart Ackermann und M. Mohr konnten 1937 in der Leber des Dornhais (Acanthias vulgaris) D-Ornithin nachweisen.[64] Die von Krebs entdeckte D-Aminosäureoxidase wurde in den folgenden Jahren in allen Säugetieren nachgewiesen. H. Blaschko und Joyce Hawkins fanden sie 1951 erstmals bei Wirbellosen. Die Funktion dieses Enzyms in den verschiedenen Organismen blieb aber weiter im Unklaren.[40] Gegen Ende der 1960er Jahre spekulierte man, dass das Enzym im Verdauungstrakt dem Abbau von Zellwandbestandteilen grampositiver Bakterien diene, die größere Mengen an D-Aminosäuren enthalten.[65] Die Theorie, dass die D-Aminosäureoxidase nur dem Abbau von außen zugeführter (exogener) D-Aminosäuren dient, hatte bis Anfang der 1990er Jahre bestand.
In der Hämolymphe der Wanzenart Oncopeltus fasciatus wurde 1950 durch Auclair und Patton[66] erstmals bei einem Vielzeller D-Alanin nachgewiesen. Sie verwendeten dabei als Analyseverfahren die zweidimensionale Papierchromatographie. Nach dem Eluieren besprühten sie die getrockneten Chromatogramme mit D-Aminosäureoxidase, die nur das D-Alanin zu einer Ketocarbonsäure desaminierte, die sich leicht mit Phenylhydrazin nachweisen ließ.[36] Als Ursache für das Vorhandensein von D-Alanin vermutete man die mikrobielle Flora, eine Aufnahme durch Nahrung, sowie eine spontane Racemisierung durch Altern.[67]
Die Biosynthese von D-Serin wurde 1965 von einer Arbeitsgruppe um John J. Corrigan an der Tufts University School of Medicine in Massachusetts nachgewiesen. Die mit radioaktiv markierter D-Glucose gefütterten Seidenspinner produzierten sowohl L- als auch D-Serin.[68] Später wurden D-Aminosäuren auch in anderen Insekten[69] und Säugetieren[70] nachgewiesen.

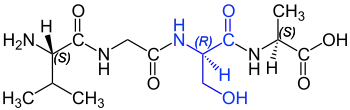
1962 isolierte eine italienische Arbeitsgruppe um Vittorio Erspamer in dem südamerikanischen Frosch der Physalaemus fuscomaculatus das Tachykinin Physalaemin.[72] Dieses Polypeptid ist aus zwölf Aminosäuren aufgebaut und beginnt, vom N-Terminus aus betrachtet, mit D-Prolin. Im Einbuchstabencode lautet die Sequenz pEADPNKFYGLM-NH2.[73] Es war das erste entdeckte natürliche Peptid mit einer D-Aminosäure, das nicht mikrobiologischen Ursprungs ist. Aber auch drei Jahre später schrieb beispielsweise der US-amerikanische Biochemiker Alton Meister in seinem Standardwerk Biochemistry of the amino acids, dass „es derzeit keine schlüssigen Beweise für das Vorkommen von D-Aminosäuren in den Proteinen von Pflanzen und Tieren gibt“.[74][22] Von Erspamers Entdeckung wurde zunächst kaum Notiz genommen. Erst als 19 Jahre später dieselbe Arbeitsgruppe in dem ebenfalls in Südamerika beheimateten Warzigen Makifrosch (Phyllomedusa sauvagii)[75][76] Dermorphin isolierte, erkannte man langsam die Tragweite der Entdeckung. Vom N-Terminus aus betrachtet hat das aus sieben Aminosäuren aufgebaute Dermorphin an Position 2 ein D-Alanin. Die D-Konfiguration des Alanins ist für die pharmakologische Aktivität unerlässlich. Dermorphin bindet an den µ1-Rezeptor und ist dabei wesentlich selektiver und potenter als die körpereigenen Endorphine (Dynorphine und Enkephaline) und das pharmakologisch weit verbreitete pflanzliche Morphin.[77] Die Entdeckung widersprach einigen Paradigmen, so dass Erspamer erhebliche Schwierigkeiten hatte, eine Fachzeitschrift zu finden, die die Ergebnisse seiner Arbeitsgruppe publizierte.[78][79]
Eines dieser Paradigmen ist, dass die DNA eines Organismus bei der Proteinbiosynthese nur für die 20 kanonischen Aminosäuren, die ausschließlich in L-Konfiguration vorliegen, codiert. Es gibt kein Gen zur Codierung von D-Aminosäuren. Dieser Widerspruch wurde über zehn Jahre später aufgelöst: Eine durch Epimerasen katalysierte stereoselektive posttranslationale Modifikation ist für das Vorkommen von D-Aminosäuren in eukaryotischen Peptiden verantwortlich. Das heißt, dass nach der Translation unter dem Einfluss eines speziellen, körpereigenen Enzyms bei einer bestimmten L-Aminosäure die Konfiguration geändert wird.[80]
D-Aminosäuren in Säugetieren
Eine biologische Funktion der D-Aminosäuren bei Säugetieren wurde bis 1992 ausgeschlossen.[67] Durch die Verbesserung von analytischen Messverfahren wie der Gas-[81] und Hochleistungsflüssigkeitschromatographie[82][83] (GC bzw. HPLC) wurde es ab den 1980er Jahren möglich, D-Aminosäuren sauber von ihren L-Spiegelbildern abzutrennen und noch in kleinsten Mengen nachzuweisen. Atsushi Hashimoto und Kollegen fanden so 1992 im Gehirn von Ratten relativ große Mengen an freiem D-Serin. Sie ermittelten eine Konzentration von etwa 0,27 µmol/g Hirnmasse. Den Gehalt an L-Serin bestimmten sie zu 0,89 µmol/g Hirnmasse, wodurch sich ein D-zu-L-Verhältnis von 0,23 ergab.[84] Schon zuvor war bekannt, dass von außen (exogen) zugeführtes D-Serin ein potenter selektiver allosterischer Agonist am NMDA-Rezeptor (N-Methyl-D-Aspartat) ist.[85] Die Quelle für die vergleichsweise hohen Konzentrationen an D-Serin, das in der Folgezeit dann auch im Gehirn anderer Säuger, einschließlich des Menschen, nachgewiesen wurde, blieb zunächst unklar. Spekulationen, wie beispielsweise die gezielte Aufnahme racemisierten L-Serins aus der Nahrung und Transport über die Blut-Hirn-Schranke in das Gehirn, endeten 1999 mit der Entdeckung des Enzyms Serinracemase im Gehirn von Ratten durch Herman Wolosker und Kollegen.[86] Serinracemase katalysiert die Racemisierung von Serin. Aminosäureracemasen kannte man zuvor nur bei Bakterien und einigen Insekten. Das Enzym wurde in Gliazellen nachgewiesen, die vergleichsweise hohe Konzentrationen an D-Serin aufweisen. Mit dem Nachweis der Serinracemase konnte gezeigt werden, dass dieser archaische D-Aminosäure-Metabolismus auch in Säugern konserviert ist[86] und – wie sich später zeigen sollte – dort eine wichtige Funktion in der Neurotransmission ausübt.[87] Das Dogma, dass D-Aminosäuren in Eukaryoten keine besonderen Funktionen haben, musste aufgegeben werden. Heute weiß man, dass D-Serin in zahlreichen Prozessen des Zentralnervensystems, wie beispielsweise Lernvorgängen und Gedächtnisfunktion, aber auch bei psychischen Erkrankungen,[88] Neuropathien und neurodegenerativen Erkrankungen,[89] eine wichtige Rolle spielt.[61][90]
Physiologische Bedeutung
Freie D-Aminosäuren
Bis zum Ende der 1990er Jahre ging man davon aus, dass D-Aminosäuren in Wirbeltieren keine physiologische Funktion haben. Mit dem Nachweis größerer Mengen von D-Serin und D-Asparaginsäure im Gehirn von Säugetieren begann die Erforschung der Funktion dieser beiden außergewöhnlichen Aminosäuren. Die Erforschung der physiologischen Wirkung der D-Aminosäuren ist eine vergleichsweise junge Disziplin mit vielen noch offenen Fragestellungen.
D-Serin
D-Serin findet sich außer in den Gliazellen auch in den Nervenzellen (Neuronen).[92][93] Es entsteht aus L-Serin unter dem katalytischen Einfluss des Enzyms Serin-Racemase (EC 5.1.1.18), das von diesen Zellen exprimiert wird. Der Abbau wird von der D-Aminosäureoxidase (EC 1.4.3.3) katalysiert. Die Konzentration an D-Serin im Gehirn wird durch diese beiden auf- beziehungsweise abbauenden Prozesse bestimmt. D-Serin wirkt als Co-Agonist am NMDA-Rezeptor, dessen „natürlicher“ Ligand die Aminosäure Glycin ist.[85] Dieser Rezeptor ist für eine Reihe von physiologischen, aber auch pathologischen Vorgängen von großer Wichtigkeit. D-Serin verstärkt die Aktivität des NMDA-Rezeptors. Es wird daher auch als ‚Neuromodulator‘ bezeichnet.[94] Eine Überexpression von D-Aminosäureoxidase, die zu einem vermehrten Abbau von D-Serin führt, reduziert folglich die Aktivität am NMDA-Rezeptor. Eine Unterfunktion der NMDA-Rezeptors wird vor allem mit Schizophrenie in Verbindung gebracht.[95] Bereits geringe Mengen von NMDA-Rezeptor-Antagonisten können bei gesunden Probanden Symptome wie kognitive und physiologische Störungen auslösen, die der einer Schizophrenie entsprechen.[96]
2002 stellte eine große internationale Arbeitsgruppe fest, dass das neu entdeckte G72-Gen (DAOA-Gen, D-amino acid oxidase activator) in engen Zusammenhang mit Schizophrenie steht. Das Genprodukt von G72 aktiviert D-Aminosäureoxidase, wodurch die Konzentration an D-Serin im Gehirn abnimmt. Zwischen der Aktivität von D-Aminosäureoxidase und dem Auftreten von Schizophrenie fanden sie nur eine schwache Korrelation. Die Kombination D-Aminosäureoxidase und G72-Aktivator war indes stark gegenseitig unterstützend (synergistisch).[97] Die Autoren schlossen daraus, dass letztlich die Konzentration an freiem D-Serin eine wesentliche Rolle bei Schizophrenie spielt. Andere Studien zeigten ebenfalls einen genetischen Zusammenhang zwischen D-Aminosäureoxidase und Schizophrenie.[98][99] Zu diesen Befunden passen die Ergebnisse von Arbeitsgruppen, die nachweisen konnten, dass die Konzentration von D-Serin im Blutserum[100] und in der Zerebrospinalflüssigkeit[101][102] von Schizophreniepatienten, im Vergleich zu einer Gruppe gesunder Probanden, signifikant reduziert ist. Darüber hinaus fand man in den Gehirnen verstorbener Schizophreniepatienten eine erhöhte Expression von D-Aminosäureoxidase.[103][104][105][106] Die zusätzliche Verabreichung von D-Serin bei der Behandlung von Patienten mit Schizophrenie zeigte in klinischen Studien vielversprechende Ergebnisse.[107][108] Bei einer Metaanalyse über 18 klinische Studien wurde eine Reduzierung der Schizophreniesymptome festgestellt. Die Verbesserung war allerdings nur moderat.[109]
Die Erkenntnisse über die Funktion von D-Serin und D-Aminosäureoxidase haben zur Entwicklung verschiedener Inhibitoren von D-Aminosäureoxidase geführt, die potenzielle Arzneimittel zur Behandlung von Schizophrenie sind.[110][111][112] Die D-Aminosäureoxidaseinhibitoren befinden sich noch in einer sehr frühen Entwicklungsphase,[113][114] so dass bisher noch kein Arzneistoff dieses Wirkprinzips zugelassen ist (Stand 2012).
Eine überhohe Konzentration dieser Aminosäure in Gliazellen und die damit einhergehende Exzitotoxizität wird als eine mögliche Ursache der amyotrophen Lateralsklerose, einer degenerativen Erkrankung des Nervensystems, untersucht.[115][116]
D-Asparaginsäure
Freie D-Asparaginsäure wurde erstmals 1986 von einer Arbeitsgruppe um den US-Amerikaner David S. Dunlop in nennenswerten Mengen im Gehirn von Nagetieren und im menschlichen Blut nachgewiesen. Dabei fanden sie in der Großhirnhemisphäre von neugeborenen Ratten mit 164 nmol/g die höchsten Konzentrationen an D-Aspartat. Dies entsprach 8,4 % der Gesamtmenge an Asparaginsäure. Dieser Konzentrationswert überschreitet denjenigen vieler essentieller L-Aminosäuren im Gehirn.[117] Außer im Gehirn konnten noch in der Zirbeldrüse, der Hypophyse, den Nebennieren und den Hoden vergleichsweise hohe Mengen an D-Aspartat nachgewiesen werden.[118][119] Analog zum D-Serin wird D-Aspartat im Organismus durch enzymatische Racemisierung von L-Aspartat, in diesem Fall durch D-Aspartatracemase (EC 5.1.1.13), gebildet und der Abbau erfolgt über D-Aspartatoxidase (EC 1.4.3.1). Die Konzentration an D-Aspartat nimmt mit zunehmendem Alter des Organismus drastisch ab.[120] Hohe Aktivitäten an D-Aspartatracemase finden sich in den Organen, in denen sich auch hohe Konzentrationen an D-Asparaginsäure nachweisen lassen. Am höchsten ist die Aktivität in der Hypophyse. Eine Deaktivierung der Aspartatracemase, beispielsweise durch Retroviren, die gezielt einen Funktionsverlust in der zur Aspartatracemase komplementären Ribonukleinsäure (RNA) hervorrufen, führt zu einer signifikanten Konzentrationsabnahme von D-Aspartat. Als Folge davon wird die dendritische Entwicklung massiv gestört, was wiederum zu ausgeprägten Schäden bei der Neurogenese im Hippocampus führt.[121] Aufgrund dieser Versuchsergebnisse geht man davon aus, dass D-Aspartat ein wichtiger Regulator der neuronalen Entwicklung ist.[120] Die genauen physiologischen Wirkungen von D-Asparaginsäure sind noch weitgehend unklar. Das Forschungsgebiet ist ausgesprochen neu. So wurde beispielsweise die Aspartatracemase erst 2010 bei Säugetieren kloniert.[121]
D-Aminosäuren-haltige Peptide
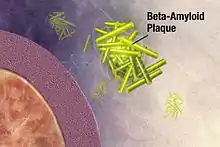
Beim Altern eines Organismus findet durch die vermehrte Racemisierung, speziell von Asparaginsäure, ein zunehmender Verlust an Homochiralität statt. Oxidativer Stress und UV-Strahlung[122] können diesen Verlust beschleunigen. Die Racemisierung von Asparaginsäure (engl. aspartic acid racemization) verläuft wegen der Bildung einer Succinimid-Zwischenstufe, die nur eine geringe Aktivierungsenergie benötigt, besonders leicht ab.[123] Diese nicht-enzymatische In-vivo-Racemisierung von Proteinen ist ein autonom ablaufender Prozess des Alterns, der vor allem langlebige Proteine wie beispielsweise das Kollagen im Dentin oder das Kristallin der Augenlinsen betrifft.[124] So racemisiert pro Lebensjahr 0,14 % der Asparaginsäure in den Augenlinsen. Bei einem 30-Jährigen sind somit durchschnittlich 4,2 % der Asparaginsäure im Kristallin der Augenlinsen racemisiert.[125] Daneben sind allerdings auch andere funktionelle Proteine, wie beispielsweise Enzyme oder Botenstoffe von der Racemisierung betroffen. Peptide, die D-Aminosäuren enthalten, sind gegenüber einem enzymatischen Abbau durch Proteasen wesentlich stabiler als Peptide, deren Aminosäuren nur in L-Konfiguration vorliegen. In vielen Fällen führt eine Racemisierung in einem körpereigenen Protein zu physiologischen Problemen. In Proteinen bewirkt die Racemisierung einen Funktionsverlust und eine Ansammlung des Proteins in den unterschiedlichsten Geweben, die der Organismus nicht mehr abbauen kann. Bei einigen Krankheitsbildern ist eine Zunahme der Racemisierung zu beobachten. Bei Arteriosklerose, Lungenemphysem, Presbyopie, grauem Star sowie Degenerationserscheinungen des Knorpels und Gehirns wird die Racemisierung von Asparaginsäure als relevanter pathologischer Faktor gesehen.[126]
1988 wurde erstmals im β-Amyloid der senilen Plaques aus dem Gehirn verstorbener Patienten mit Alzheimer-Krankheit ein erhöhter Racemisierungsgrad festgestellt.[127] Vor allem D-Aspartat und D-Serin konnten nachgewiesen werden.[128] Später wurde erkannt, dass eine Racemisierung der Asparaginsäure in Position 23[129] zu einer beschleunigten Peptidaggregation führt,[130] die als ein wesentliches Element bei der Pathogenese der Alzheimer-Krankheit gesehen wird. Im Gegensatz zur Racemisierung in Position 23 führt die Racemisierung in Position 7 zu einer verminderten Peptidaggregation.[130] Den vermutlich durch Proteinalterung hervorgerufenen Racemisierungsprozessen des β-Amyloids, die ähnlich denen beim Dentin verlaufen, wird eine wichtige Rolle bei der Entstehung der Alzheimer-Krankheit zugeschrieben. Die Racemisierung beschleunigt die Peptidaggregation und erschwert den enzymatischen Abbau durch Proteasen.[131][132]
Eigenschaften
Chemische und physikalische Eigenschaften
In einem achiralen Umfeld sind D- und L-Aminosäuren in ihren chemischen und physikalischen Eigenschaften, mit Ausnahme der Drehrichtung von polarisiertem Licht, völlig gleich. In einem chiralen Umfeld können erhebliche Unterschiede festgestellt werden. Dies gilt insbesondere bei biochemischen Prozessen, die von Natur aus chiral sind. Ein praktisches Beispiel hierfür ist der Geschmacksunterschied zwischen den Aminosäure-Enantiomeren. Die aus L-Aminosäuren aufgebauten G-Protein-gekoppelten Geschmacksrezeptoren sind ein chirales Umfeld, mit dem Enantiomere unterschiedlich wechselwirken. So wird der Geschmack der meisten L-Aminosäuren als ‚bitter‘ beschrieben, während der von D-Aminosäuren meist als ‚süß‘ bezeichnet wird.[133][134] Ein extremes Beispiel ist dabei D-Tryptophan; die mit Abstand süßeste Aminosäure hat die 37-fache Süßkraft wie Saccharose. L-Tryptophan ist dagegen zusammen mit L-Tyrosin die bitterste Aminosäure.[1] Entsprechend unterschiedlich können auch die Wechselwirkungen mit anderen Rezeptoren oder Enzymen bei biochemischen Vorgängen ausfallen. Dies gilt insbesondere auch für Peptide und Proteine, die eine oder mehrere D-Aminosäuren enthalten.
Der Einbau einer D- beziehungsweise die Epimerisierung einer L-Aminosäure in einem Protein bewirkt aus stereochemischer Sicht die Bildung eines Diastereomers, das dem gesamten Protein völlig neue chemische und physikalische Eigenschaften verleiht.[32] Biochemisch hat dieser Eingriff in die Primärstruktur erhebliche Auswirkungen auf die daraus abgeleitete Sekundär-, Tertiär- und Quartärstruktur des Peptids. Die biochemische Wirkung wird dabei stark verändert. Sie kann in den beiden Extremfällen entweder völlig verloren gehen (loss of function) oder völlig neuartige, beispielsweise toxische Wirkungen zur Folge haben (gain of function). D-Aminosäuren verhindern in einem sonst aus L-Aminosäuren aufgebauten Peptid, dass eine α-Helix ausgebildet werden kann. Sie sind ‚helixbrechend‘. Nur Proteine, die vollständig aus D- oder L-Aminosäuren aufgebaut sind, können – wenn helixbildende Aminosäuren wie Valin, Glutamin, Isoleucin, Alanin, Methionin, Leucin, Glutaminsäure oder Tryptophan vorhanden sind – eine Helixstruktur ausbilden, die spiegelbildlich zueinander sind. Bei gemischt aufgebauten Peptiden ist dies nicht möglich.[15]
Toxikologie
D-Isomere proteinogener Aminosäuren

In Studien, in denen die extensive orale Aufnahme von Aminosäuren – beispielsweise in Form von Nahrungsergänzungsmitteln – untersucht wurde, zeigten, mit Ausnahme von Serin und Asparaginsäure, alle Aminosäuren in der „natürlichen“ L-Konfiguration stärker toxische Effekte als das entsprechende D-Enantiomer.[135][27] D-Aminosäuren sind ein natürlicher Bestandteil einer Vielzahl von Lebensmitteln. Sie entstehen dort vor allem durch Racemisierungsprozesse aus den „natürlichen“ L-Aminosäuren. In Lebensmitteln, die einen Fermentierungsprozess durchlaufen haben, wie beispielsweise Milchprodukte, finden sich erhöhte Mengen an D-Aminosäuren. So enthält Emmentaler etwa 0,7 g/kg an D-Aminosäuren.[136] Schon im Ausgangsprodukt Kuhmilch liegen etwa 1,5 % aller Aminosäuren in der D-Konfiguration vor.[137]
Es wird geschätzt, dass etwa ein Drittel der über die Nahrung aufgenommenen D-Aminosäuren mikrobiellen Ursprungs ist.[138] Um die in der Nahrung enthaltenen und in Proteinen verknüpften Aminosäuren für den Organismus nutzen zu können, müssen die Proteine während der Verdauung in ihre Einzelbestandteile, die freien Aminosäuren, zerlegt werden. Befinden sich in einem Protein D-Aminosäuren, so kann die Zugänglichkeit des Proteins für proteolytische Enzyme erheblich eingeschränkt sein. Die Enzyme des menschlichen Verdauungsapparates können keine Bindungen zwischen D- und L-Aminosäuren spalten. Der Abbau in einzelne Aminosäuren, Di- oder Tripeptide, der notwendig ist, um über die Darmschleimhäute vom Organismus aufgenommen werden zu können,[139] ist dadurch erschwert. Größere Peptidbruchstücke können nicht verwertet werden und werden mit dem Fäzes ausgeschieden. Die Bioverfügbarkeit, und damit auch der Nährwert, ist dann stark vermindert.[140] Di- oder Tripeptide, die D-Aminosäuren enthalten, können – wie auch freie D-Aminosäuren – über Peptidtransporter resorbiert werden. Ein Großteil der so aufgenommenen D-Aminosäuren wird über die Nieren wieder ausgeschieden. Abhängig vom Nahrungsangebot und der jeweiligen D-Aminosäure wird ein Teil der D-Aminosäuren per Transaminierung in L-Aminosäuren umgewandelt und so der Proteinbiosynthese zugänglich gemacht.[141]
Der Einbau von „unnatürlichen“ D-Aminosäuren in die Zellwand von Bakterien bewirkt deren Beständigkeit gegen Proteasen. Diese Proteasestabilität ist auch für den Menschen von großer Wichtigkeit, schließlich befinden sich im Darm eines Erwachsenen mehrere hundert Gramm Darmbakterien, die zusammen mit einer Vielzahl von Proteasen für die Verdauung unerlässlich sind.
Der größte Teil an D-Aminosäuren in der Nahrung entsteht bei deren Zubereitung. Hohe Temperaturen und stark saure oder basische Bedingungen führen zur (Teil)-Racemisierung. Beispielsweise liegen in Kartoffelchips etwa 14 % der Asparaginsäure in der D-Form vor. In Kaffeeweißer sind es 17 % und in Frühstücksspeckstreifen 13 %. Freie L-Aminosäuren racemisieren etwa zehn Mal langsamer als proteingebundene. Der Racemisierungsgrad ist außerdem von der Aminosäure selbst stark abhängig. So neigt Serin, bedingt durch die Hydroxygruppe, besonders leicht zur Racemisierung.[142][140][143] Die bei der Produktion von Gelatine benötigten drastischen Bedingungen – entweder saurer oder basischer Aufschluss bei erhöhten Temperaturen – führen zu einer starken Racemisierung, speziell der Asparaginsäure, im Kollagen der Gelatine. Der Anteil an D-Aspartat am Gesamt-Aspartat kann bei kommerziell erhältlicher Gelatine leicht über 30 % liegen.[144]
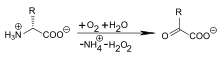
D-Aminosäuren werden bei ihrer Aufnahme durch den Säugetierorganismus nicht in Proteine oder Peptide oder andere (Makro)-Moleküle des Stoffwechsels eingebaut. Eine Anreicherung im Körpergewebe in unveränderter Form ist nicht zu beobachten. Über die Nahrung oder per Infusion aufgenommene D-Aminosäuren werden zum Teil über den Urin ausgeschieden und zum Teil über das in Leber und Nieren vorhandene Enzym D-Aminosäureoxidase per Desaminierung in die „normalen“ Stoffwechselprodukte, die Ketocarbonsäuren, oxidiert. Bezüglich der Toxizität von infundierten D-Aminosäuren liegen, mehr oder weniger unfreiwillig, langjährige Erfahrungen vor, die darauf schließen lassen, dass D-Aminosäuren nicht gesundheitsschädlich sind.[145] Die Basis dieser Aussage ist die gute Verträglichkeit der parenteralen Nahrung („künstlichen Ernährung“), die lange Jahre aus hochdosierten Aminosäure-Racematen bestand. Diese Infusionslösungen wurden mittels saurer Hydrolyse – die zwangsläufig zur Racemisierung führt – aus Proteinen hergestellt.[146] Racemisches Methionin (DL-Methionin) ist Bestandteil vieler Futtermittel in der Viehwirtschaft. In Milchkühen konnte nachgewiesen werden, dass über 75 % des D-Methionins in L-Methionin transformiert und dadurch bioverfügbar wird.[147]
Unabhängig von diesen Erfahrungswerten sind Versuchsergebnisse am Tiermodell Ratte zu sehen. Hohe Dosen (im Bereich von 0,8 g/kg Körpergewicht) an D-Serin führen in diesen Modellorganismen zu einer akuten tubulären Nekrose,[148][149][150][151] die nach Absetzen der D-Serin-Gabe reversibel ist.[152] Nach etwa sechs Tagen ist die vollständige Regeneration der Nierenfunktion abgeschlossen.[153] Die pathologischen Veränderungen ähneln weitgehend der einer durch Lysinoalanin ausgelösten Nierenschädigung. Warum D-Serin in diesen hohen Konzentrationen nierentoxisch ist, ist noch nicht sicher geklärt. Möglicherweise reduziert das D-Serin die Konzentration an renalem Glutathion, das die proximalen Tubuluszellen vor den schädlichen Einflüssen reaktiver Sauerstoffspezies (ROS) schützen soll. Beim enzymatischen Abbau von D-Serin durch D-Aminosäureoxidase entsteht als Nebenprodukt Wasserstoffperoxid,[154] das den intrazellulären Vorrat an Glutathion deutlich herabsetzt.[155][140]

Für großes Aufsehen sorgte im Dezember 1989 eine in der angesehenen Fachzeitschrift The Lancet veröffentlichte Mitteilung dreier Ärzte aus Wien. Sie hatten in Milch, die sie per Mikrowellengerät erhitzten, größere Mengen an D-Prolin gefunden, das offensichtlich durch Racemisierung von L-Prolin entstanden war. Des Weiteren schrieben sie dem D-Prolin neuro-, nephro- und hepatotoxische Eigenschaften zu.[156] Bei der Veröffentlichung handelte es sich um einen Letter an die Herausgeber, und nicht um eine Peer-Review-Veröffentlichung oder gar eine kontrollierte Studie.[157] Auch nannten die Autoren nicht die Versuchsbedingungen, unter denen dieser Racemisierungsgrad erreicht wurde.
Unabhängig davon wurde die Meldung von der Tages- und Wochenpresse mit dramatisierenden Formulierungen und Warnungen vor der Verwendung von Mikrowellengeräten publiziert. Im August 1990 gab es vom Bundesgesundheitsamt eine Klarstellung des Sachverhaltes, die allerdings kaum öffentliche Wirkung zeigte. Andere Wissenschaftler wiesen darauf hin, dass D-Prolin ein normaler Bestandteil der täglichen Nahrung ist, der nach der oralen Aufnahme schnell abgebaut und ausgeschieden wird.[158] Dennoch erschien beispielsweise im August 1991 eine Zeitschrift mit der Schlagzeile „Mikrowellen vergiften Nerven, Leber und Nieren“.[146] Ähnliche Behauptungen finden sich heute noch auf einschlägigen Websites.[159][160]
Versuche anderer Arbeitsgruppen, die Ergebnisse der Wiener Ärzte zu reproduzieren, schlugen zunächst fehl. So konnte auch bei einem 30-minütigen Kochen von Milch auf der Herdplatte keine Zunahme an D-Prolin gemessen werden.[161][162] Zwei Jahre später wurden die Versuchsbedingungen veröffentlicht. Die Autoren des Lancet-Letters hatten die Milch in einem geschlossenen Druckgefäß für 10 Minuten auf 174 bis 176 °C erhitzt – ein Temperaturbereich, der in haushaltsüblichen Gefäßen zur Milcherhitzung nicht erreicht werden kann.[163][164]
Bei ihrer Aussage zur Neurotoxizität von D-Prolin bezogen sich die Autoren des Lancet-Letters auf Versuche aus dem Jahr 1978, bei denen Hühnerküken die Substanz intraventrikulär, das heißt direkt in ein Hirnventrikel, injiziert wurde.[165]
Spätere Untersuchungen zur Toxizität von D-Prolin bei Ratten zeigten, dass die Verbindung auch in hohen Konzentrationen ungefährlich ist.[166][146]
Eine reale Gefahr beim Erhitzen von Milch mittels Mikrowellengerät geht – speziell für Kleinkinder – von der ungleichmäßigen Erwärmung des Flascheninhaltes aus, die häufig zu klinisch relevanten Verbrennungen führt.[167]
D-Isomere nicht-proteinogener Aminosäuren
Über die Toxizität der D-Isomere nicht-proteinogener Aminosäuren lassen sich keine allgemeinen Aussagen treffen. Sie ist von Aminosäure zu Aminosäure sehr individuell. Interessanterweise sind einige Verbindungen, die D-Aminosäuren enthalten, deutlich weniger toxisch als ihre L-Isomeren. Beispiele hierfür sind Cycloserin und Penicillamin. So liegt beispielsweise der LD50-Wert für die orale Gabe des Racemates aus D- und L-Penicillamin im Modellorganismus Ratte bei 365 mg/kg. Für das reine D-Penicillamin sind auch bei einer Dosis von 1200 mg/kg dagegen keinerlei Anzeichen einer Toxizität gegeben.[168]
D-Peptide
Allgemeine Aussagen über die toxikologischen Eigenschaften von D-Peptiden sind nicht möglich. Die Empfindlichkeit gegenüber Proteasen ist deutlich geringer und das immunogene Potenzial ist signifikant niedriger als bei den entsprechenden L-Peptiden.[169][170]
Analyse
Klassische Verfahren

Mit einem Polarimeter lässt sich der optische Drehwinkel einer Aminosäurenlösung bestimmen, aus dem der Gehalt an D- und L-Enantiomeren errechnet werden kann. Dazu sind jedoch standardisierte Bedingungen (vor allem Konzentration, Temperatur und Lösungsmittel) notwendig. Zudem ist das Verfahren nur für einzelne Aminosäuren und nicht für Gemische unterschiedlicher Aminosäuren geeignet. In den 1960er bis 1980er Jahre wurde auch die Ionenaustauschchromatographie zur Auftrennung derivatisierter Aminosäuren verwendet. Dabei wurden die zu analysierenden Aminosäuren vor der Auftrennung mit L-Aminosäuren zu diastereomeren Dipeptiden umgesetzt.[171] Auch enzymatische Verfahren, die auf der Umsetzung mit spezifischen Enzymen, wie L- und D-Aminosäureoxidase basieren,[172] gehören zu den klassischen Verfahren der Enantiomerenbestimmung von Aminosäuren.[27] Als nicht-chromatographisches Verfahren ist unter anderem auch die Kapillarelektrophorese zur Analyse von D-Aminosäuren geeignet.[27]
Chromatographische Verfahren
Quantitative Analysen, auch komplexer Gemische von Aminosäuren, lassen sich mit Hilfe von chromatographischen Verfahren durchführen. Dabei werden zunächst die einzelnen Komponenten des Gemisches an einer stationären Phase aufgetrennt und anschließend mit einem Detektor gemessen. Als Detektoren kommen vor allem UV- oder Massenspektrometer, in der Gaschromatographie auch Flammenionisationsdetektoren, zum Einsatz. Zur Trennung des Ausgangsgemisches an der stationären Phase werden zwei unterschiedliche Strategien angewendet. Im einfachsten Fall erfolgt die Auftrennung der beiden Enantiomeren an einer chiralen stationären Phase, mit der die beiden Isomere unterschiedlich stark wechselwirken und somit unterschiedlich schnell eluieren. An einer achiralen stationären Phase ist die Trennung nur möglich, wenn die Enantiomere in Diastereomere überführt werden. Als Analysenverfahren haben sich vor allem die Gaschromatographie (GC) und die Hochleistungsflüssigkeitschromatographie (HPLC) etabliert. Die Enantiomerenreinheit von D-Aminosäuren kann auch dünnschichtchromatographisch analysiert werden.[173]
Erst die Entwicklung spezieller chromatographischer Methoden ermöglichte den Nachweis und die Quantifizierung von D-Aminosäuren in den Organen höherer Organismen.[174][175]
Gaschromatographie

Aminosäuren können nicht zersetzungsfrei verdampft werden. Für die Auftrennung und Analyse in der Gaschromatographie müssen sie in unzersetzt verdampfbare Verbindungen überführt werden. Dazu werden die Aminosäuren meist einem zweistufigen Derivatisierungsprozess unterzogen. So kann beispielsweise im ersten Schritt die Veresterung der Carboxygruppe mit Ethanol und danach, in einem zweiten Schritt, die Umsetzung der Aminogruppe mit Trifluoressigsäureanhydrid zum Trifluoracetyl-Derivat (TFA) erfolgen. Das dabei gebildete N-TFA/O-Ethyl-Derivat der Aminosäure kann im Gaschromatographen unzersetzt verdampft und an einer chiralen stationären Phase getrennt werden.[176] Die Derivatisierung mit chiralen Reagenzien birgt die erhöhte Gefahr einer Racemisierung, und dass die Reaktionspartner unterschiedliche Reaktionskinetiken aufweisen. Beides kann das Messergebnis verfälschen.[27]
Hochleistungsflüssigkeitschromatographie

In der HPLC hat sich im Vergleich zur Gaschromatographie die Derivatisierung mit chiralen Reagenzien und der Einsatz nicht-chiraler stationärer Phasen, beispielsweise RP-18, durchgesetzt. Zur Derivatisierung wird beispielsweise L-N-Acetylcystein zusammen mit Phthaldialdehyd verwendet.[177] Das dabei entstehende Diastereomerenpaar (D-L und L-L) hat unterschiedliche chemische und physikalische Eigenschaften, wodurch es dann auf einer konventionellen Säule getrennt und anschließend detektiert werden kann.
Synthese

Die meisten proteinogenen L-Aminosäuren werden fermentativ hergestellt. Für D-Aminosäuren ist dieses mikrobiologische Verfahren nicht geeignet.[178] Um den zunehmenden Bedarf an D-Aminosäuren zu decken, wurden verschiedene Produktionsverfahren entwickelt.
Die klassischen chemischen Synthesen, wie beispielsweise die Strecker-Synthese, liefern stets die Racemate der Aminosäuren. Aus diesen Gemischen können die einzelnen Aminosäuren entweder aufwändig abgetrennt werden (Racematspaltung) oder man setzt die L-Aminosäure enzymatisch mittels L-Aminosäuredesaminasen zur Ketocarbonsäure um, die sich vergleichsweise leicht abtrennen lässt.[179][180]
Eleganter ist die D-Aminosäuresynthese über substituierte Hydantoine. Hydantoine lassen sich großtechnisch nach der Bucherer-Bergs-Reaktion (auch Bucherer-Bergs-Hydantoinsynthese genannt) aus Aldehyden, Kaliumcyanid und Ammoniumcarbonat produzieren. Über die Wahl des eingesetzten Aldehyds wird die entstehende Aminosäure bestimmt. Das so produzierte Hydantoin kann im sogenannten Hydantoinase-Verfahren zur D-Aminosäure weiter umgesetzt werden. Dieses Multi-Enzymverfahren wurde von der Degussa (heute Evonik Degussa) entwickelt und besteht aus drei Reaktionsschritten. Zunächst wird das racemische Hydantoin-Derivat unter dem katalytischen Einfluss einer D-Hydantoinase zur N-Carbamoyl-D-Aminosäure hydrolysiert. Im zweiten Schritt wird die N-Carbamoyl-D-Aminosäure mit Hilfe einer D-Carbamoylase zur enantiomerenreinen Aminosäure weiter hydrolysiert. Im dritten Schritt wird das bei der Synthese nicht umgesetzte Enantiomer des Hydantoin-Derivates chemisch oder enzymatisch racemisiert. Die chemische Racemisierung erfolgt bei pH-Werten >8 und kann durch den Zusatz einer Racemase deutlich beschleunigt werden. Im Vergleich zu anderen Verfahren werden beim Hydantoinase-Verfahren ausgehend vom Racemat enantiomerenreine Aminosäuren mit theoretischen Ausbeuten von bis zu 100 % produziert.[181]
Verwendung
Der weltweite Bedarf an D-Aminosäuren ist über die letzten Jahre kontinuierlich gestiegen. Für das Jahr 2017 wird eine Marktgröße von etwa 3,7 Mrd. US-Dollar prognostiziert.[182]
D-Aminosäuren finden sich als wichtige Bausteine beispielsweise in Süßstoffen, in Insektiziden, in Kosmetika und vor allem in einer Vielzahl von peptidischen Arzneimitteln, die ein wesentlicher Wachstumstreiber für die Marktentwicklung sind.[183]
So werden jährlich mehrere tausend Tonnen an D-4-Hydroxyphenylglycin und D-Phenylglycin zur Synthese von Penicillinen (beispielsweise Amoxicillin) und Cephalosporinen (beispielsweise Cefaclor) benötigt.[184]
D-Aminosäuren erhöhen nicht nur in den Zellwänden von Bakterien die Stabilität gegenüber einem proteolytischen Abbau, auch der gezielte Einbau in Arzneimittel erhöht deren Stabilität, speziell bei der oralen Einnahme. Die Änderung der Anordnung der funktionellen Gruppen (Konformation) bietet zudem beim Molekülaufbau einen weiteren Freiheitsgrad in der Gestaltung der Molekülstruktur, die zu verbesserten Wirkstoffeigenschaften führen kann.[184] Der in der Reproduktionsmedizin eingesetzte Gonadorelin-Inhibitor Cetrorelix, ein GnRH-Analogon, besteht beispielsweise aus zehn Aminosäuren, von denen fünf in der D-Konfiguration vorliegen.[185][186] Cetrorelix wird vollsynthetisch aus den einzelnen Aminosäuren aufgebaut. Auch andere GnRH-Analoga wie Leuprorelin, Buserelin, Degarelix, Histrelin, Nafarelin oder Abarelix enthalten mindestens eine D-Aminosäure.
Das zur Behandlung der erektilen Dysfunktion verwendete Tadalafil, besser bekannt unter dem Markennamen Cialis, wird bei der Synthese aus D-Tryptophan aufgebaut.[187] Das Antidiabetikum Nateglinid, aus der Gruppe der Glinide, wird aus D-Phenylalanin und cis-4-Isopropyl-cyclohexan-carbonsäure hergestellt. Phenylalanin wird seit den 1970er Jahren als Antidepressivum verwendet.[188] Zum Einsatz als Arzneimittel kommt das kostengünstige Racemat. Ein wesentlicher Teil der antidepressiven und schmerzstillenden Wirkung geht dabei vom D-Phenylalanin aus, das – im Vergleich zum L-Phenylalanin – nicht zu stimmungsverbesserndem L-Tyrosin, L-DOPA oder Noradrenalin verstoffwechselt wird, sondern primär das Enzym Enkephalinase hemmt.[189] Durch die Blockade der Enkephalinase wird der Blutspiegel an Enkephalinen im Blut erhöht, was den ebenfalls zu beobachtenden schmerzstillenden Effekt hervorruft. Im weiteren Verlauf wird das D-Phenylalanin dann vor allem zu Phenylethylamin verstoffwechselt.[190][191]
Das unter anderem zur Bekämpfung der Varroamilbe zugelassene Insektizid Fluvalinat[192] aus der Gruppe der Pyrethroide wird aus D-Valin hergestellt.
D-Alanin ist ein Bestandteil des Süßstoffes Alitam.
Weiterführende Literatur
- Ryuichi Konno, Hans Brückner, Antimo D'Aniello, George Fisher, Noriko Fujii, Hiroshi Homma: D-amino acids: a new frontier in amino acids and protein research – practical methods and protocols. Nova Science Publishers, 2007, ISBN 1-60021-075-9, 629 S.
- Loredano Pollegioni, Stefano Servi (Hrsg.): Unnatural Amino Acids. Humana Press, 2011, ISBN 1-61779-330-2, 409 S.
- Gyula Pályi, Luciano Caglioti, Claudia Zucchi (Hrsg.): Advances in BioChirality. Elsevier, 1999, ISBN 0-08-043404-5 (eingeschränkte Vorschau in der Google-Buchsuche).
Weblinks
- Die D- und die L-Form der Aminosäuren
- Hanka Symmank: Funktionelle und strukturelle Charakterisierung bakterieller Peptidsynthetasen. Fachbereich Biologie, Chemie, Pharmazie, Freie Universität Berlin, Mai 2002
Einzelnachweise
- Hans-Dieter Belitz, Werner Grosch, Peter Schieberle: Lehrbuch der Lebensmittelchemie. 5. Auflage, Springer Verlag, 2001. ISBN 3-540-41096-1 (eingeschränkte Vorschau in der Google-Buchsuche).
- Uwe Meierhenrich: Amino Acids and the Asymmetry of Life: Caught in the Act of Formation. Springer, 2008, ISBN 3-540-76885-8, S. 53–54 (eingeschränkte Vorschau in der Google-Buchsuche).
- V. S. Lamzin, Z. Dauter, K. S. Wilson: How nature deals with stereoisomers. In: Current opinion in structural biology. Band 5, Nummer 6, Dezember 1995, S. 830–836, PMID 8749373. (Review).
- S. A. Fuchs, R. Berger u. a.: D-amino acids in the central nervous system in health and disease. In: Molecular Genetics and Metabolism. Band 85, Nummer 3, Juli 2005, S. 168–180, doi:10.1016/j.ymgme.2005.03.003. PMID 15979028. (Review).
- G. F. Joyce, G. M. Visser u. a.: Chiral selection in poly(C)-directed synthesis of oligo(G). In: Nature. Band 310, Nummer 5978, 1984 Aug 16-22, S. 602–604, PMID 6462250.
- V. V. Avetisov, V. I. Goldanskii: Homochirality and stereospecific activity: evolutionary aspects. In: Bio Systems. Band 25, Nummer 3, 1991, S. 141–149, PMID 1912384.
- N. Fujii, T. Saito: Homochirality and life. In: Chemical record. Band 4, Nummer 5, 2004, S. 267–278, doi:10.1002/tcr.20020. PMID 15543607. (Review).
- W. A. Bonner: Experimental evidence for beta-decay as a source of chirality by enantiomer analysis. In: Origins of life. Band 14, Nummer 1–4, 1984, S. 383–390, PMID 11536584. (Review).
- W. A. Bonner: Parity violation and the evolution of biomolecular homochirality. In: Chirality. Band 12, Nummer 3, März 2000, S. 114–126, doi:10.1002/(SICI)1520-636X(2000)12:3<114::AID-CHIR3>3.0.CO;2-N. PMID 10689289. (Review).
- J. R. Cronin, S. Pizzarello: Enantiomeric excesses in meteoritic amino acids. In: Science. Band 275, Nummer 5302, Februar 1997, S. 951–955, PMID 9020072.
- S. Pizzarello, M. Zolensky, K. A. Turk: Nonracemic isovaline in the Murchison meteorite: chiral distribution and mineral association. In: Geochimica et Cosmochimica Acta. Band 67, Nummer 8, 2003, S. 1589–1595. doi:10.1016/S0016-7037(02)01283-8.
- P. Schmitt-Kopplin, Z. Gabelica u. a.: High molecular diversity of extraterrestrial organic matter in Murchison meteorite revealed 40 years after its fall. In: PNAS. Band 107, Nummer 7, Februar 2010, S. 2763–2768, doi:10.1073/pnas.0912157107. PMID 20160129. PMC 2840304 (freier Volltext).
- D. P. Glavin, J. P. Dworkin: Enrichment of the amino acid L-isovaline by aqueous alteration on CI and CM meteorite parent bodies. In: PNAS. Band 106, Nummer 14, April 2009, S. 5487–5492, doi:10.1073/pnas.0811618106. PMID 19289826. PMC 2667035 (freier Volltext).
- P. W. Lucas, J. H. Hough u. a.: UV circular polarisation in star formation regions: the origin of homochirality? In: Origins of life and evolution of the biosphere. Band 35, Nummer 1, Februar 2005, S. 29–60, PMID 15889649.
- T. Carell: (Seite nicht mehr abrufbar, Suche in Webarchiven: Vorlesung Stereochemie.) Kapitel 9: Racemisierungen LMU München, S. 150.
- Elizabeth R. Neswald: Thermodynamik als kultureller Kampfplatz: zur Faszinationsgeschichte der Entropie, 1850–1915. Rombach, 2003, ISBN 3-7930-9448-0, S. 335.
- A. S. Kekulé: Ach wie gut, dass niemand weiß … In: Tagesspiegel. Vom 12. Januar 2011.
- T. Ogino, H. Ogino: Application to forensic odontology of aspartic acid racemization in unerupted and supernumerary teeth. In: Journal of dental research. Band 67, Nummer 10, Oktober 1988, S. 1319–1322, PMID 3170888.
- T. Ogino, H. Ogino, B. Nagy: Application of aspartic acid racemization to forensic odontology: post mortem designation of age at death. In: Forensic science international. Band 29, Nummer 3–4, 1985, S. 259–267, PMID 4076954.
- S. Ohtani, T. Yamamoto: Strategy for the estimation of chronological age using the aspartic acid racemization method with special reference to coefficient of correlation between D/L ratios and ages. In: Journal of forensic sciences. Band 50, Nummer 5, 2005, S. 1020–1027, PMID 16225206. (Review).
- J. L. Bada, B. Herrmann u. a.: Amino acid racemization in bone and the boiling of the German Emperor Lothar I. In: Applied Geochemistry. Band 4, Nummer 3, 1989, S. 325–327, doi:10.1016/0883-2927(89)90036-X.
- Chris McManus: Right Hand, Left Hand – The Origins of Asymmetry in Brains, Bodies, Atoms and Cultures. Harvard University Press, 2004, ISBN 0-674-01613-0, S. 130 (eingeschränkte Vorschau in der Google-Buchsuche).
- P. M. Masters, M. Friedman: Racemization of amino acids in alkali-treated food proteins. In: Journal of agricultural and food chemistry. Band 27, Nummer 3, 1979 May-Jun, S. 507–511, PMID 447924.
- J. L. Bada: Kinetics of racemization of amino acids as a function of pH. In: Journal of the American Chemical Society. Band 94, Nummer 4, Februar 1972, S. 1371–1373, PMID 5060280.
- H. Frank, W. Woiwode u. a.: Determination of the rate of acidic catalyzed racemization of protein amino acids. In: Liebigs Ann Chem. Nummer 3, 1981, S. 354–365. doi:10.1002/jlac.198119810303.
- T. Geiger, S. Clarke: Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. In: The Journal of biological chemistry. Band 262, Nummer 2, Januar 1987, S. 785–794, PMID 3805008.
- Thorsten Erbe: Die Quantifizierung von Aminosäurenisomeren in Lebensmitteln mittels chiraler Gaschromatographie-Massenspektrometrie im Hinblick auf die Relevanz und die Entstehungsmechanismen von D-Aminosäuren. Dissertation, Justus-Liebig-Universität Gießen, 1999.
- A. Paquet, M. Ching-Yung: Racemization assessment in alkali treated dietary proteins using high-performance liquid chromatography. In: Nutrition Research. Band 9, Nummer 9, 1989, S. 1053–1065. doi:10.1016/S0271-5317(89)80066-1.
- M. Friedman: Chemistry, nutrition, and microbiology of D-amino acids. In: Journal of agricultural and food chemistry. Band 47, Nummer 9, September 1999, S. 3457–3479, PMID 10552672. (Review).
- J. P. Richard, T. L. Amyes: Proton transfer at carbon. In: Current opinion in chemical biology. Band 5, Nummer 6, Dezember 2001, S. 626–633, PMID 11738171. (Review).
- J. P. Richard, T. L. Amyes: On the importance of being zwitterionic: enzymatic catalysis of decarboxylation and deprotonation of cationic carbon. In: Bioorganic Chemistry. Band 32, Nummer 5, Oktober 2004, S. 354–366, doi:10.1016/j.bioorg.2004.05.002. PMID 15381401. (Review).
- Daniel Björn Stein: Substratspezifität und Funktionalität von Epimerisierungsdomänen in der nichtribosomalen Peptidsynthese. Dissertation, Philipps-Universität Marburg, 2006, S. 29.
- S. Glavas, M. E. Tanner: Active site residues of glutamate racemase. In: Biochemistry. Band 40, Nummer 21, Mai 2001, S. 6199–6204, PMID 11371180.
- L. M. Fisher, J. G. Belasco u. a.: Energetics of proline racemase: transition-state fractionation factors for the two protons involved in the catalytic steps. In: Biochemistry. Band 25, Nummer 9, Mai 1986, S. 2543–2551, PMID 3521738.
- Geoffrey Zubay: Origins of Life: On Earth and in the Cosmos. Academic Press, 2000, ISBN 0-12-781910-X, S. 296 (eingeschränkte Vorschau in der Google-Buchsuche).
- J. J. Corrigan: D-amino acids in animals. In: Science. Band 164, Nummer 3876, April 1969, S. 142–149, PMID 5774186.
- E. Abderhalden: Fermentforschung. Band 16–17, S. Hirzel, 1942, S. 301.
- H. A. Krebs: Untersuchungen über den Stoffwechsel der Aminosäuren im Tierkörper. In: Hoppe-Seyler's Zeitschrift für physiologische Chemie. Band 217, 1933, S. 191.
- H. A. Krebs: Metabolism of amino-acids: Deamination of amino-acids. In: The Biochemical journal. Band 29, Nummer 7, Juli 1935, S. 1620–1644, PMID 16745832. PMC 1266672 (freier Volltext).
- H. Blaschko, J. Hawkins: D-Amino acid oxidase in the molluscan liver. In: The Biochemical journal. Band 52, Nummer 2, Oktober 1952, S. 306–310, PMID 13018226. PMC 1197987 (freier Volltext).
- F. Ehrlich: Über asymmetrische und symmetrische Einwirkung von Hefe auf Racemverbindungen natürlich vorkommender Aminosäuren. In: Biochem Z. 63, 1914, S. 379–401.
- E. O. von Lippmann: Ueber das Vorkommen von Leucin und Tyrosin in der Rübenmelasse. In: Ber Dtsch Chem Ges. Band 17, 1994, S. 2835–2840. doi:10.1002/cber.188401702243.
- S. Fraenkel, H. Gallia, A. Liebster, S. Rosen: Über die Produkte prolongierter tryptischer Verdauung des Caseins. In: Biochem Z. Band 145, 1924, S. 225–241.
- E. Winterstein, C. Reuter und R. Korolew: Ueber die chemische Zusammensetzung einiger Pilze und über die bei der Autolyse derselben auftretenden Produkte. In: Landw Versuchsstat. LXXIX - LXX, 1913, S. 541–562.
- J. H. Birkinshaw, H. Raistrick, G. Smith: Studies in the biochemistry of micro-organisms: Fumaryl-dl-alanine (fumaromono-dl-alanide), a metabolic product of Penicillium resticulosum sp.nov. In: The Biochemical journal. Band 36, Nummer 10–12, Dezember 1942, S. 829–835, PMID 16747516. PMC 1266878 (freier Volltext).
- T. Robinson: D-amino acids in higher plants. In: Life sciences. Band 19, Nummer 8, Oktober 1976, S. 1097–1102, PMID 792607. (Review).
- J. L. Frahn, R. J. Illman: The occurrence of D-alanine and D-alanyl-D-alanine in Phalaris tuberosa. In: Phytochem Band 14, 1975, S. 1464–1465. doi:10.1016/S0031-9422(00)98674-6.
- Y. Gogami, K. Ito u. a.: Occurrence of D-serine in rice and characterization of rice serine racemase. In: Phytochemistry. Band 70, Nummer 3, Februar 2009, S. 380–387, doi:10.1016/j.phytochem.2009.01.003. PMID 19249065.
- T. Ogawa, M. Fukuda, K. Sasaoka: Occurrence of N-malonyl-D-alanine in pea seedlings. In: Biochimica et Biophysica Acta. Band 297, Nummer 1, Januar 1973, S. 60–69, PMID 4144329.
- H. Brückner, S. Haasmann, A. Friedrich: Quantification of D-amino acids in human urine using GC-MS and HPLC. In: Amino Acids. Band 6, 1994, S. 205–211. doi:10.1007/BF00805848.
- E. E. Snell, B. M. Guirard: Some Interrelationships of Pyridoxine, Alanine and Glycine in Their Effect on Certain Lactic Acid Bacteria. In: PNAS. Band 29, Nummer 2, 1943, S. 66–73, PMID 16588604. PMC 1078561 (freier Volltext).
- J. Olivard, E. E. Snell: Growth and enzymatic activities of vitamin B6 analogues. I. D-Alanine synthesis. In: The Journal of biological chemistry. Band 213, Nummer 1, März 1955, S. 203–214, PMID 14353919. PMC 1078561 (freier Volltext).
- E. E. Snell: The vitamin B6 group: VII. Replacement of vitamin B6 for some microorganisms by d(−)-alanine and an unidentified factor from casein. In: J Biol Chem. Band 158, 1945, S. 497–503.
- Albert Gossauer: Struktur und Reaktivität der Biomoleküle. John Wiley & Sons, 2003, ISBN 3-906390-29-2, S. 347 (eingeschränkte Vorschau in der Google-Buchsuche).
- W. A. Wood, I. C. Gunsalus: D-Alanine formation; a racemase in Streptococcus faecalis. In: The Journal of biological chemistry. Band 190, Nummer 1, Mai 1951, S. 403–416, PMID 14841188.
- J. Ju, H. Misono, K. Ohnishi: Directed evolution of bacterial alanine racemases with higher expression level. In: Journal of bioscience and bioengineering. Band 100, Nummer 3, September 2005, S. 246–254, doi:10.1263/jbb.100.246. PMID 16243272.
- R. J. Thompson, H. G. Bouwer u. a.: Pathogenicity and immunogenicity of a Listeria monocytogenes strain that requires D-alanine for growth. In: Infection and immunity. Band 66, Nummer 8, August 1998, S. 3552–3561, PMID 9673233. PMC 108386 (freier Volltext).
- D. Billot-Klein, L. Gutmann u. a.: Modification of peptidoglycan precursors is a common feature of the low-level vancomycin-resistant VANB-type Enterococcus D366 and of the naturally glycopeptide-resistant species Lactobacillus casei, Pediococcus pentosaceus, Leuconostoc mesenteroides, and Enterococcus gallinarum. In: Journal of bacteriology. Band 176, Nummer 8, April 1994, S. 2398–2405, PMID 8157610. PMC 205365 (freier Volltext).
- P. E. Reynolds, H. A. Snaith u. a.: Analysis of peptidoglycan precursors in vancomycin-resistant Enterococcus gallinarum BM4174. In: The Biochemical journal. Band 301, Juli 1994, S. 5–8, PMID 8037690. PMC 1137133 (freier Volltext).
- C. A. Arias, M. Martín-Martinez u. a.: Characterization and modelling of VanT: a novel, membrane-bound, serine racemase from vancomycin-resistant Enterococcus gallinarum BM4174. In: Molecular microbiology. Band 31, Nummer 6, März 1999, S. 1653–1664, PMID 10209740.
- Norma Christine Stäbler: Untersuchungen zur Bildung von D-Aminosäuren mit Corynebacterium glutamicum. Dissertation, Heinrich-Heine-Universität Düsseldorf, 2010, S. 7.
- M. F. Freeman, C. Gurgui u. a.: Metagenome Mining Reveals Polytheonamides as Posttranslationally Modified Ribosomal Peptides. In: Science. [elektronische Veröffentlichung vor dem Druck] September 2012, doi:10.1126/science.1226121. PMID 22983711.
- T. Hamada, S. Matsunaga u. a.: Solution structure of polytheonamide B, a highly cytotoxic nonribosomal polypeptide from marine sponge. In: Journal of the American Chemical Society. Band 132, Nummer 37, September 2010, S. 12941–12945, doi:10.1021/ja104616z. PMID 20795624.
- D. Ackermann, M. Mohr: Über stickstoffhaltige Bestandteile der Leber des Haifisches (Acanthias vulgaris). In: Z Biol. Band 98, Nummer 37, 1937, S. 26.
- L. R. Lyle, J. W. Jutila: D-amino acid oxidase induction in the kidneys of germ-free mice. In: Journal of bacteriology. Band 96, Nummer 3, September 1968, S. 606–608, PMID 4389707. PMC 252348 (freier Volltext).
- J. L. Auclair, R. L. Patton: On the occurrence of D-Alanine in the haemolymph of the milkweed bug, Oncopeltus fasciatus. In: Revue canadienne de biologie. Band 9, Nummer 1, April 1950, S. 3–8, PMID 15417891.
- Gianluca Molla, Luciano Piubelli u. a.: Enzymatic Detection of D-Amino Acids. In: Loredano Pollegioni, Stefano Servi (Hrsg.): Unnatural Amino Acids. Band 794, 2012, ISBN 978-1-61779-330-1, S. 273–289. doi:10.1007/978-1-61779-331-8_18.
- N. G. Srinivasan, J. J. Corrigan, A. Meister: Biosynthesis of D-Serine in the silkworm, Bombyx mori. (PDF; 665 kB) In: The Journal of biological chemistry. Band 240, Februar 1965, S. 796–800, PMID 14275137.
- J. J. Corrigan, N. G. Srinivasan: The occurrence of certain D-amino acids in insects. In: Biochemistry. Band 5, Nummer 4, April 1966, S. 1185–1190, PMID 5958195.
- Y. Nagata, K. Yamamoto u. a.: The presence of free D-alanine, D-proline and D-serine in mice. In: Biochimica et Biophysica Acta. Band 1115, Nummer 3, Januar 1992, S. 208–211, PMID 1346751.
- P. Melchiorri, L. Negri: The dermorphin peptide family. In: General pharmacology. Band 27, Nummer 7, Oktober 1996, S. 1099–1107, PMID 8981054. (Review).
- A. Anastasi, V. Erspamer, J. M. Cei: Isolatioan and amino acid sequence of physalaemin, the main active polypeptide of the skin of Physalaemus fuscumaculatus. In: Archives of biochemistry and biophysics. Band 108, November 1964, S. 341–348, PMID 14240587.
- Rebecca Jo Jackway: Biologically Active Peptides from Australian Amphibians. PhD Thesis, University of Adelaide, 2008, S. 165.
- wörtlich: At this time there is no conclusive evidence for the occurance of D-amino acids in the proteins of plants and animals. Alton Meister: Biochemistry of the amino acids Academic Press, 1965.
- M. Broccardo, V. Erspamer u. a.: Pharmacological data on dermorphins, a new class of potent opioid peptides from amphibian skin. In: British journal of pharmacology. Band 73, Nummer 3, Juli 1981, S. 625–631, PMID 7195758. PMC 2071698 (freier Volltext).
- V. Erspamer, P. Melchiorri u. a.: Deltorphins: a family of naturally occurring peptides with high affinity and selectivity for delta opioid binding sites. In: PNAS. Band 86, Nummer 13, Juli 1989, S. 5188–5192, PMID 2544892. PMC 297583 (freier Volltext).
- M. Amiche, A. Delfour, P. Nicolas: Opiod peptides from frog skin. In: Pierre Jollès (Hrsg.): D-Amino Acids in Sequences of Secreted Peptides of Multicellular Organisms. Springer, 1998, ISBN 3-7643-5814-9, S. 57–72 (eingeschränkte Vorschau in der Google-Buchsuche).
- L. H. Lazarus, M. Attila: The toad, ugly and venomous, wears yet a precious jewel in his skin. In: Progress in neurobiology. Band 41, Nummer 4, Oktober 1993, S. 473–507, PMID 8210414. (Review).
- G. Kreil: Peptides containing a D-amino acid from frogs and molluscs. In: The Journal of biological chemistry. Band 269, Nummer 15, April 1994, S. 10967–10970, PMID 8157620. (Review).
- S. D. Heck, W. S. Faraci u. a.: Posttranslational amino acid epimerization: enzyme-catalyzed isomerization of amino acid residues in peptide chains. In: PNAS. Band 93, Nummer 9, April 1996, S. 4036–4039, PMID 8633012. PMC 39482 (freier Volltext).
- R. Liardon, R. Jost: Racemization of free and protein-bound amino acids in strong mineral acid. In: International journal of peptide and protein research. Band 18, Nummer 5, November 1981, S. 500–505, PMID 7341532.
- H. Brückner, T. Westhauser, H. Godel: Liquid chromatographic determination of D- and L-amino acids by derivatization with o-phthaldialdehyde and N-isobutyryl-L-cysteine. Applications with reference to the analysis of peptidic antibiotics, toxins, drugs and pharmaceutically used amino acids. In: Journal of chromatography. A. Band 711, Nummer 1, September 1995, S. 201–215, PMID 7496491.
- R. H. Buck, K. Krummen: High-performance liquid chromatographic determination of enantiomeric amino acids and amino alcohols after derivatization with o-phthaldialdehyde and various chiral mercaptans. Application to peptide hydrolysates. In: Journal of chromatography. Band 387, Januar 1987, S. 255–265, PMID 3558624.
- A. Hashimoto, T. Nishikawa u. a.: The presence of free D-serine in rat brain. In: FEBS letters. Band 296, Nummer 1, Januar 1992, S. 33–36, PMID 1730289.
- N. W. Kleckner, R. Dingledine: Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. In: Science. Band 241, Nummer 4867, August 1988, S. 835–837, PMID 2841759.
- H. Wolosker, S. Blackshaw, S. H. Snyder: Serine racemase: a glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. In: PNAS. Band 96, Nummer 23, November 1999, S. 13409–13414, PMID 10557334. PMC 23961 (freier Volltext).
- H. Wolosker, E. Dumin u. a.: D-amino acids in the brain: D-serine in neurotransmission and neurodegeneration. In: The FEBS journal. Band 275, Nummer 14, Juli 2008, S. 3514–3526, doi:10.1111/j.1742-4658.2008.06515.x. PMID 18564180. (Review).
- S. Sacchi, M. Bernasconi u. a.: pLG72 modulates intracellular D-serine levels through its interaction with D-amino acid oxidase: effect on schizophrenia susceptibility. In: The Journal of biological chemistry. Band 283, Nummer 32, 2008, S. 22244–22256, doi:10.1074/jbc.M709153200. PMID 18544534.
- H. J. Ryu, J. E. Kim u. a.: Potential roles of D-serine and serine racemase in experimental temporal lobe epilepsy. In: Journal of neuroscience research. Band 88, Nummer 11, 2010, S. 2469–2482, doi:10.1002/jnr.22415. PMID 20623543.
- S. A. Fuchs, R. Berger, T. J. de Koning: D-serine: the right or wrong isoform? In: Brain research. Band 1401, Juli 2011, S. 104–117, doi:10.1016/j.brainres.2011.05.039. PMID 21676380. (Review).
- Julia Scharlau: Untersuchungen zur Auswirkung von adoleszenter chronischer Cannabinoidbehandlung an einem Mausmodell der Schizophrenie. Dissertation, Friedrich-Wilhelms-Universität Bonn, 2012, urn:nbn:de:hbz:5n-28406. S. 16.
- E. Kartvelishvily, M. Shleper u. a.: Neuron-derived D-serine release provides a novel means to activate N-methyl-D-aspartate receptors. In: The Journal of biological chemistry. Band 281, Nummer 20, Mai 2006, S. 14151–14162, doi:10.1074/jbc.M512927200. PMID 16551623.
- K. Miya, R. Inoue u. a.: Serine racemase is predominantly localized in neurons in mouse brain. In: The Journal of comparative neurology. Band 510, Nummer 6, Oktober 2008, S. 641–654, doi:10.1002/cne.21822. PMID 18698599.
- L. Pollegioni, S. Sacchi: Metabolism of the neuromodulator D-serine. In: Cellular and molecular life sciences. Band 67, Nummer 14, Juli 2010, S. 2387–2404, doi:10.1007/s00018-010-0307-9. PMID 20195697. (Review).
- J. T. Kantrowitz, D. C. Javitt: N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? In: Brain research bulletin. Band 83, Nummer 3–4, September 2010, S. 108–121, doi:10.1016/j.brainresbull.2010.04.006. PMID 20417696. PMC 2941541 (freier Volltext). (Review).
- J. T. Coyle: Glutamate and schizophrenia: beyond the dopamine hypothesis. In: Cellular and molecular neurobiology. Band 26, Nummer 4–6, 2006 Jul-Aug, S. 365–384, doi:10.1007/s10571-006-9062-8. PMID 16773445. (Review).
- I. Chumakov, M. Blumenfeld u. a.: Genetic and physiological data implicating the new human gene G72 and the gene for D-amino acid oxidase in schizophrenia. In: PNAS. Band 99, Nummer 21, Oktober 2002, S. 13675–13680, doi:10.1073/pnas.182412499. PMID 12364586. PMC 129739 (freier Volltext).
- A. Corvin, K. A. McGhee u. a.: Evidence for association and epistasis at the DAOA/G30 and D-amino acid oxidase loci in an Irish schizophrenia sample. In: American journal of medical genetics. Band 144B, Nummer 7, Oktober 2007, S. 949–953, doi:10.1002/ajmg.b.30452. PMID 17492767.
- T. Ohnuma, N. Shibata u. a.: Association analysis of glycine- and serine-related genes in a Japanese population of patients with schizophrenia. In: Progress in neuro-psychopharmacology & biological psychiatry. Band 33, Nummer 3, April 2009, S. 511–518, doi:10.1016/j.pnpbp.2009.02.004. PMID 19223009.
- K. Hashimoto, T. Fukushima u. a.: Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. In: Archives of general psychiatry. Band 60, Nummer 6, Juni 2003, S. 572–576, doi:10.1001/archpsyc.60.6.572. PMID 12796220.
- I. Bendikov, C. Nadri u. a.: A CSF and postmortem brain study of D-serine metabolic parameters in schizophrenia. In: Schizophrenia research. Band 90, Nummer 1–3, Februar 2007, S. 41–51, doi:10.1016/j.schres.2006.10.010. PMID 17156977.
- K. Hashimoto, G. Engberg u. a.: Reduced D-serine to total serine ratio in the cerebrospinal fluid of drug naive schizophrenic patients. In: Progress in neuro-psychopharmacology & biological psychiatry. Band 29, Nummer 5, Juni 2005, S. 767–769, doi:10.1016/j.pnpbp.2005.04.023. PMID 15939521.
- L. Verrall, M. Walker u. a.: d-Amino acid oxidase and serine racemase in human brain: normal distribution and altered expression in schizophrenia. In: The European journal of neuroscience. Band 26, Nummer 6, September 2007, S. 1657–1669, doi:10.1111/j.1460-9568.2007.05769.x. PMID 17880399. PMC 2121142 (freier Volltext).
- L. Verrall, P. W. Burnet u. a.: The neurobiology of D-amino acid oxidase and its involvement in schizophrenia. In: Molecular psychiatry. Band 15, Nummer 2, Februar 2010, S. 122–137, doi:10.1038/mp.2009.99. PMID 19786963. PMC 2811712 (freier Volltext). (Review).
- R. Kapoor, K. S. Lim u. a.: Preliminary evidence for a link between schizophrenia and NMDA-glycine site receptor ligand metabolic enzymes, d-amino acid oxidase (DAAO) and kynurenine aminotransferase-1 (KAT-1). In: Brain research. Band 1106, Nummer 1, August 2006, S. 205–210, doi:10.1016/j.brainres.2006.05.082. PMID 16828464.
- C. Madeira, M. E. Freitas u. a.: Increased brain D-amino acid oxidase (DAAO) activity in schizophrenia. In: Schizophrenia research. Band 101, Nummer 1–3, April 2008, S. 76–83, doi:10.1016/j.schres.2008.02.002. PMID 18378121.
- J. T. Coyle, G. Tsai, D. C. Goff: Ionotropic glutamate receptors as therapeutic targets in schizophrenia. In: Current drug targets. Band 1, Nummer 2, April 2002, S. 183–189, PMID 12769626. (Review).
- G. Tsai, P. Yang u. a.: D-serine added to antipsychotics for the treatment of schizophrenia. In: Biological psychiatry. Band 44, Nummer 11, Dezember 1998, S. 1081–1089, PMID 9836012.
- H. J. Tuominen, J. Tiihonen, K. Wahlbeck: Glutamatergic drugs for schizophrenia. In: Cochrane database of systematic reviews (Online). Nummer 2, 2006, S. CD003730, doi:10.1002/14651858.CD003730.pub2. PMID 16625590. (Review).
- D. V. Ferraris, T. Tsukamoto: Recent advances in the discovery of D-amino acid oxidase inhibitors and their therapeutic utility in schizophrenia. In: Current pharmaceutical design. Band 17, Nummer 2, 2011, S. 103–111, PMID 21361869. (Review).
- C. A. Strick, C. Li u. a.: Modulation of NMDA receptor function by inhibition of D-amino acid oxidase in rodent brain. In: Neuropharmacology. Band 61, Nummer 5–6, 2011 Oct-Nov, S. 1001–1015, doi:10.1016/j.neuropharm.2011.06.029. PMID 21763704.
- T. Sparey, P. Abeywickrema u. a.: The discovery of fused pyrrole carboxylic acids as novel, potent D-amino acid oxidase (DAO) inhibitors. In: Bioorganic & medicinal chemistry letters. Band 18, Nummer 11, Juni 2008, S. 3386–3391, doi:10.1016/j.bmcl.2008.04.020. PMID 18455394.
- Berry JF, Ferraris DV, Duvall B, et al.: Synthesis and SAR of 1-hydroxy-1H-benzo[d]imidazol-2(3H)-ones as Inhibitors of D-Amino Acid Oxidase. In: ACS Med Chem Lett. 3, Nr. 10, 2012, S. 839–843. doi:10.1021/ml300212a. PMID 23243487.
- Sacchi S, Rosini E, Pollegioni L, Molla G: D-Amino Acid Oxidase Inhibitors as a Novel Class of Drugs for Schizophrenia Therapy. In: Curr. Pharm. Des.. 2012. PMID 23116391.
- Paul P, de Belleroche J: The role of D-amino acids in amyotrophic lateral sclerosis pathogenesis: a review. In: Amino Acids. 43, Nr. 5, 2012, S. 1823–1831. doi:10.1007/s00726-012-1385-9. PMID 22890612.
- Sasabe J, Chiba T, Yamada M, et al.: D-serine is a key determinant of glutamate toxicity in amyotrophic lateral sclerosis. In: EMBO J.. 26, Nr. 18, 2007, S. 4149–4159. doi:10.1038/sj.emboj.7601840. PMID 17762863. PMC 2230675 (freier Volltext).
- D. S. Dunlop, A. Neidle u. a.: The presence of free D-aspartic acid in rodents and man. In: Biochemical and biophysical research communications. Band 141, Nummer 1, November 1986, S. 27–32, PMID 3801000.
- H. Wolosker, A. D'Aniello, S. H. Snyder: D-aspartate disposition in neuronal and endocrine tissues: ontogeny, biosynthesis and release. In: Neuroscience. Band 100, Nummer 1, 2000, S. 183–189, PMID 10996468.
- M. Katane, H. Homma: D-aspartate oxidase: the sole catabolic enzyme acting on free D-aspartate in mammals. In: Chemistry & biodiversity. Band 7, Nummer 6, Juni 2010, S. 1435–1449, doi:10.1002/cbdv.200900250. PMID 20564562. (Review).
- H. Ohide, Y. Miyoshi u. a.: D-Amino acid metabolism in mammals: biosynthesis, degradation and analytical aspects of the metabolic study. In: Journal of chromatography. B Band 879, Nummer 29, November 2011, S. 3162–3168, doi:10.1016/j.jchromb.2011.06.028. PMID 21757409. (Review).
- P. M. Kim, X. Duan u. a.: Aspartate racemase, generating neuronal D-aspartate, regulates adult neurogenesis. In: PNAS. Band 107, Nummer 7, Februar 2010, S. 3175–3179, doi:10.1073/pnas.0914706107. PMID 20133766. PMC 2840285 (freier Volltext).
- Y. Mori, K. Aki u. a.: UV B-irradiation enhances the racemization and isomerizaiton of aspartyl residues and production of N?-carboxymethyl lysine (CML) in keratin of skin. In: Journal of chromatography B. Band 879, Nummer 29, November 2011, S. 3303–3309, doi:10.1016/j.jchromb.2011.05.010. PMID 21636332.
- N. Fujii, Y. Kaji u. a.: Collapse of homochirality of amino acids in proteins from various tissues during aging. In: Chemistry & biodiversity. Band 7, Nummer 6, Juni 2010, S. 1389–1397, doi:10.1002/cbdv.200900337. PMID 20564552. (Review).
- N. Fujii: D-amino acid in elderly tissues. In: Biological & pharmaceutical bulletin. Band 28, Nummer 9, September 2005, S. 1585–1589, PMID 16141520. (Review).
- Graham C. Barrett, Donald Trevor Elmore: Amino Acids and Peptides. Cambridge University Press, 1998, ISBN 0-521-46292-4, S. 15–16 (eingeschränkte Vorschau in der Google-Buchsuche).
- S. Ritz-Timme, M. J. Collins: Racemization of aspartic acid in human proteins. In: Ageing research reviews. Band 1, Nummer 1, Februar 2002, S. 43–59, PMID 12039448. (Review).
- H. Mori, K. Ishii u. a.: Racemization: its biological significance on neuropathogenesis of Alzheimer's disease. In: The Tohoku journal of experimental medicine. Band 174, Nummer 3, November 1994, S. 251–262, PMID 7761990.
- R. Shapira, G. E. Austin, S. S. Mirra: Neuritic plaque amyloid in Alzheimer's disease is highly racemized. In: Journal of neurochemistry. Band 50, Nummer 1, Januar 1988, S. 69–74, PMID 3121789.
- A. E. Roher, J. D. Lowenson u. a.: Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer's disease. In: The Journal of biological chemistry. Band 268, Nummer 5, Februar 1993, S. 3072–3083, PMID 8428986.
- T. Tomiyama, S. Asano u. a.: Racemization of Asp23 residue affects the aggregation properties of Alzheimer amyloid beta protein analogues. In: The Journal of biological chemistry. Band 269, Nummer 14, April 1994, S. 10205–10208, PMID 8144598.
- Patrick R. Hof, Charles V. Mobbs: Handbook of the Neuroscience of Aging. Academic Press, 2009, ISBN 0-12-374898-4, S. 41 (eingeschränkte Vorschau in der Google-Buchsuche).
- M. L. Moro, M. J. Collins, E. Cappellini: Alzheimer's disease and amyloid beta-peptide deposition in the brain: a matter of 'aging'? In: Biochemical Society transactions. Band 38, Nummer 2, April 2010, S. 539–544, doi:10.1042/BST0380539. PMID 20298218. (Review).
- Günter Westphal, Gerhard Gerber, Bodo Lipke: Proteine – Nutritive und funktionelle Eigenschaften. Springer, 2003 ISBN 3-540-00232-4, S. 125 (eingeschränkte Vorschau in der Google-Buchsuche).
- Gerhard G. Habermehl, Peter E. Hammann u. a.: Naturstoffchemie. 3. Auflage, Springer, 2008, ISBN 3-540-73732-4, S. 252 (eingeschränkte Vorschau in der Google-Buchsuche).
- N. J. Benevenga, R. D. Steele: Adverse effects of excessive consumption of amino acids. In: Annual review of nutrition. Band 4, 1984, S. 157–181, doi:10.1146/annurev.nu.04.070184.001105. PMID 6235826. (Review).
- J. Zagon, L.-I. Dehne, K.-W. Bögl: D-Amino acids in organisms and food. In: Nutr Res. Band 14, 1994, S. 445–463.
- Werner Baltes, Reinhard Matissek: Lebensmittelchemie 7. Auflage, Springer, 2011, ISBN 3-642-16538-9, S. 164 (eingeschränkte Vorschau in der Google-Buchsuche).
- W. Leuchtenberger, K. Huthmacher, K. Drauz: Biotechnological production of amino acids and derivatives: current status and prospects. In: Applied Microbiology and Biotechnology. Band 69, Nummer 1, November 2005, S. 1–8, doi:10.1007/s00253-005-0155-y. PMID 16195792. (Review).
- F. H. Leibach, V. Ganapathy: Peptide transporters in the intestine and the kidney. In: Annual review of nutrition. Band 16, 1996, S. 99–119, doi:10.1146/annurev.nu.16.070196.000531. PMID 8839921. (Review).
- M. Friedman: Origin, microbiology, nutrition, and pharmacology of D-amino acids. In: Chemistry & biodiversity. Band 7, Nummer 6, 2010, S. 1491–1530, doi:10.1002/cbdv.200900225. PMID 20564567. (Review).
- W. Heine, K. Wutzke, U. Drescher: D-amino acid utilization in infants measured with the 15N-tracer technique. In: Clinical nutrition. Band 2, Nummer 1, April 1983, S. 31–35, PMID 16829405.
- M. Friedman, R. Liardon: Racemization kinetics of amino acid residues in alkali-treated soybean proteins. In: J Agric Food Chem. Band 33, 1985, S. 666–672.
- J. C. Crawhall, D. F. Elliott: A note on the racemization of serine. In: The Biochemical journal. Band 48, Nummer 2, Februar 1951, S. 237–238, PMID 14820833. PMC 1275510 (freier Volltext).
- M. Lüpke, H. Brückner: Gas chromatographic evaluation of amino acid epimerisation in the course of gelatin manufacturing and processing. In: Zeitschrift für Lebensmitteluntersuchung und -Forschung A. Band 206, Nummer 5, 1998, S. 323–328. doi:10.1007/s002170050266.
- Ernährungsbericht 1996. Deutsche Gesellschaft für Ernährung, 1996, ISBN 3-921606-33-0, S. 149.
- Johannes Friedrich Diehl: Chemie in Lebensmitteln. John Wiley & Sons, 2012, ISBN 3-527-66084-4, S. 95 (eingeschränkte Vorschau in der Google-Buchsuche).
- H. Lapierre, G. Holtrop u. a.: Is D-methionine bioavailable to the dairy cow? In: Journal of dairy science. Band 95, Nummer 1, Januar 2012, S. 353–362, doi:10.3168/jds.2011-4553. PMID 22192214.
- W. H. Fishman, C. Artom: Serine injury. In: J Biol Chem. Band 145, 1942, S. 345–346.
- R. P. Morehead, C. Artom, W. H. Fishman: The nephrotoxic action of serine. In: Proceedings. American Federation for Clinical Research. Band 2, 1945, S. 81, PMID 20275626.
- F. A. Carone, C. E. Ganote: D-serine nephrotoxicity. The nature of proteinuria, glucosuria, and aminoaciduria in acute tubular necrosis. In: Archives of pathology. Band 99, Nummer 12, Dezember 1975, S. 658–662, PMID 1203037.
- R. E. Williams, E. A. Lock: D-serine-induced nephrotoxicity: possible interaction with tyrosine metabolism. In: Toxicology. Band 201, Nummer 1–3, September 2004, S. 231–238, doi:10.1016/j.tox.2004.05.001. PMID 15297036.
- D. R. Peterson, F. A. Carone: Renal regeneration following d-serine induced acute tubular necrosis. In: The Anatomical record. Band 193, Nummer 3, März 1979, S. 383–388, doi:10.1002/ar.1091930305. PMID 426302.
- C. E. Ganote, D. R. Peterson, F. A. Carone: The nature of D-serine–induced nephrotoxicity. In: The American journal of pathology. Band 77, Nummer 2, November 1974, S. 269–282, PMID 4447130. PMC 1910915 (freier Volltext).
- M. S. Pilone, L. Pollegioni: D-amino acid oxidase as an industrial biocatalyst. In: Biocatalysis and Biotransformation. Band 20, Nummer 3, 2002, S. 145–159. doi:10.1080/10242420290020679.
- A. W. Krug, K. Völker u. a.: Why is D-serine nephrotoxic and alpha-aminoisobutyric acid protective? (Memento des Originals vom 16. Januar 2016 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. In: American Journal of Physiology - Renal physiology. Band 293, Nummer 1, 2007, S. F382–F390, doi:10.1152/ajprenal.00441.2006. PMID 17429029.
- G. Lubec, C. Wolf, B. Bartosch: Aminoacid isomerisation and microwave exposure. In: Lancet. Band 334, Nummer 8676, Dezember 1989, S. 1392–1393, PMID 2574327.
- Karl S. Kruszelnicki: Microwaves damage food. In: ABC abcscience. Vom 23. März 2006
- W. Segal: Microwave heating of milk. In: Lancet. Band 335, Nummer 8687, Februar 1990, S. 470, PMID 1968186.
- Die Wirkung von mit Mikrowellen bestrahltem Wasser auf Pflanzen. Abgerufen am 24. August 2012.
- Gefährliche Mikrowellen. Vom 24. Oktober 2008, abgerufen am 28. September 2012.
- P. Fritz, L. I. Dehne, J. Zagon, K. W. Bögl: Zur Frage der Aminosäureisomerisierung im Mikrowellenfeld: Ergebnisse eines Modellversuches mit Standardlösungen. In: Zeitschrift für Ernährungswissenschaft. Band 31, Nummer 3, 1992, S. 219–224.
- L. I. Dehne, P. Fritz: Isomerisierung von Aminosäuren durch Mikrowellenerhitzung? In: Bundesgesundheitsblatt. Band 35, 1992, S. 463–464.
- P. Sieber, P. Eberhardt, P. U. Gallmann: Heat treatment of milk in domestic microwave ovens. In: Int Dairy Journal. Band 6, 1996, S. 231–246, doi:10.1016/0958-6946(95)00009-7.
- J. Zagon, L. Dehne, K.-W. Bögl: Isomerisierung von Aminosäuren in Lebensmitteln. Teil I: Mechanismen und Verbreitung der Aminosäurenisomerisierung in Organismen und Lebensmitteln. In: Ernährungs-Umschau. Band 38, 1991, S. 275–278 und 324–328.
- A. Cherkin, J. L. Davis, M. W. Garman: D-Proline: stereospecificity and sodium chloride dependence of lethal convulsant activity in the chick. In: Pharmacology, biochemistry, and behavior. Band 8, Nummer 5, Mai 1978, S. 623–625, PMID 674269.
- A. Schieber, H. Brückner u. a.: Evaluation of D-amino acid levels in rat by gas chromatography-selected ion monitoring mass spectrometry: no evidence for subacute toxicity of orally fed D-proline and D-aspartic acid. In: Journal of chromatography. Band 691, Nummer 1, März 1997, S. 1–12, PMID 9140753.
- S. N. Ali, G. O'Toole, M. Tyler: Milk bottle burns. In: The Journal of burn care & rehabilitation. Band 25, Nummer 5, 2004, S. 461–462, PMID 15353942.
- I. A. Jaffe, K. Altman, P. Merryman: The antipyridoxine effect of penicillamine in man. In: The Journal of clinical investigation. Band 43, Oktober 1964, S. 1869–1873, doi:10.1172/JCI105060. PMID 14236210. PMC 289631 (freier Volltext).
- T. N. Schumacher, L. M. Mayr u. a.: Identification of D-peptide ligands through mirror-image phage display. In: Science. Band 271, Nummer 5257, März 1996, S. 1854–1857, PMID 8596952.
- Katja Wiesehan: Identifizierung und Charakterisierung eines spezifischen Liganden für das Alzheimer Amyloid-β-Peptid (Aβ). (PDF; 11,1 MB) Dissertation, Universität Bayreuth, 2003, S. 21.
- J. M. Manning, S. Moore: Determination of D- and L-amino acids by ion exchange chromatography as L-D and L-L dipeptides. In: The Journal of biological chemistry. Band 243, Nummer 21, November 1968, S. 5591–5597, PMID 5699053.
- K. Soda: Microdetermination of D-amino acids and D-amino acid oxidase activity with 3,methyl-2-benzothiazolone hydrazone hydrochloride. In: Analytical biochemistry. Band 25, Nummer 1, Oktober 1968, S. 228–235, PMID 4387536.
- doi:10.1002/ardp.19863190515.
- D. L. Kirschner, T. K. Green: Separation and sensitive detection of D-amino acids in biological matrices. In: Journal of separation science. Band 32, Nummer 13, Juli 2009, S. 2305–2318, doi:10.1002/jssc.200900101. PMID 19569111. (Review).
- K. Hamase, A. Morikawa u. a.: Analysis of small amounts of D-amino acids and the study of their physiological functions in mammals. In: Analytical sciences. Band 25, Nummer 8, August 2009, S. 961–968, PMID 19667471. (Review).
- Clarissa Dorothee Marzini: Grenzen der quantitativen Enantiomeranalytik von alpha-Aminosäuren. (PDF; 1,7 MB) Dissertation, Eberhard-Karls-Universität Tübingen, 2007, S. 20–21.
- N. Nimura, T. Kinoshita: O-Phthaldialdehyde N-acetyl-L-cysteine as a chiral derivatization reagent for liquid chromatographic optical resolution of amino acid enantiomers and ist application to conventional amino acid analysis. In: Journal of Chromatography A. Band 352, 1986, S. 169–177. doi:10.1016/S0021-9673(01)83377-X.
- Birgit Geueke: Neue Aminosäureoxidasen aus Rhodococcus opacus und Arthrobacter protophormiae: Untersuchungen zur biochemischen Charakterisierung, Klonierung und Expression. (PDF; 9,1 MB) Dissertation, Heinrich-Heine-Universität Düsseldorf, 2002, S. 1.
- E. Takahashi, M. Furui u. a.: D-lysine production from L-lysine by successive chemical racemization and microbial asymmetric degradation. In: Applied Microbiology and Biotechnology. Band 47, Nummer 4, April 1997, S. 347–351, PMID 9163947.
- K. Isobe, H. Tamauchi u. a.: A Simple Enzymatic Method for Production of a Wide Variety of D-Amino Acids Using L-Amino Acid Oxidase from Rhodococcus sp. AIU Z-35-1. In: Enzyme research. Band 2010, 2010, S. 567210, doi:10.4061/2010/567210. PMID 21048866. PMC 2962901 (freier Volltext).
- Kerstin Ragnitz: Immobilisierung und Stabilisierung der Hydantoinase und L-N-Carbamoylase aus Arthrobacter aurescens DSM 3747. (PDF; 997 kB) Dissertation, Universität Stuttgart, 2000, S. 4.
- Global D-Amino Acids Market to Reach $3.7 Billion by 2017, According to a New Report by Global Industry Analysts, Inc. Bei: prweb.com vom 25. August 2011.
- S. Martínez-Rodríguez, A. I. Martínez-Gómez u. a.: Natural occurrence and industrial applications of D-amino acids: an overview. In: Chemistry & biodiversity. Band 7, Nummer 6, Juni 2010, S. 1531–1548, doi:10.1002/cbdv.200900245. PMID 20564568. (Review).
- Markus Werner: Klonierung der D-Carbamoylase aus Arthrobacter crystallopoietes DSM 20117. Dissertation, Universität Stuttgart, 2001.
- B. Kutscher, M. Bernd: Chemie und Molekularbiologie bei der Suche nach neuen LHRH-Antagonisten. In: Angew Chem. Band 109, Nummer 20, 1997, S. 2240–2254, doi:10.1002/ange.19971092005.
- T. Beckers, M. Bernd u. a.: Structure-function studies of linear and cyclized peptide antagonists of the GnRH receptor. In: Biochemical and biophysical research communications. Band 289, Nummer 3, Dezember 2001, S. 653–663, doi:10.1006/bbrc.2001.5939. PMID 11726197.
- Alaa A.-M. Abdel-Aziz, Yousif A. Asiri u. a.: Tadalafil. (Memento des Originals vom 24. Dezember 2012 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. (PDF; 4,4 MB) In: Harry G. Brittain (Hrsg.): Profiles of Drug Substances, Excipients and Related Methodology. Academic Press, 2011, ISBN 0-12-387702-4, S. 287–329 (eingeschränkte Vorschau in der Google-Buchsuche).
- E. Fischer, B. Heller u. a.: Therapy of depression by phenylalanine. Preliminary note. In: Arzneimittel-Forschung. Band 25, Nummer 1, Januar 1975, S. 132, PMID 1173765.
- S. Ehrenpreis: D-phenylalanine and other enkephalinase inhibitors as pharmacological agents: implications for some important therapeutic application. In: Substance and alcohol actions/misuse. Band 3, Nummer 4, 1982, S. 231–239, PMID 6301083.
- Mitchell Bebel Stargrove, Jonathan Treasure, Dwight L. McKee: Herb, Nutrient, and Drug Interactions. Elsevier Health Sciences, 2008, ISBN 0-323-02964-7, S. 682 (eingeschränkte Vorschau in der Google-Buchsuche).
- Phenylalanine. University of Maryland Medical Center, abgerufen am 21. September 2012.
- D. L. Williams: A veterinary approach to the European honey bee (Apis mellifera) In: Veterinary journal. Band 160, Nummer 1, Juli 2000, S. 61–73, doi:10.1053/tvjl.2000.0474. PMID 10950136. (Review).