Klonierung
Klonierung (oder Klonieren, engl. molecular cloning) ist in der Molekularbiologie der Überbegriff für Methoden zur Gewinnung und identischen Vervielfältigung von Desoxyribonukleinsäure (DNA). Im Gegensatz zum Klonen, dessen Ziel in der Herstellung genetisch identischer Organismen besteht, beschränkt sich die Klonierung auf die Herstellung identischer Moleküle der DNA.
Eigenschaften
Bei der Klonierung wird ein gewünschtes DNA-Fragment (z. B. ein Gen oder die für ein Protein codierende cDNA) in einen Vektor (z. B. ein Plasmid oder viraler Vektor) integriert. Das Ziel einer Klonierung ist, ein DNA-Fragment zu vermehren, um seine Eigenschaften zu untersuchen oder in weiteren Prozessen weiterzuverwenden. Nach einer Vervielfältigung kann durch Isolierung der Plasmid-DNA ein Vielfaches der anfangs eingesetzten DNA-Menge gewonnen werden, was im Gegensatz zu in vitro-Verfahren wie der PCR kostengünstig, präzise und in großer Zahl geschieht. Alternativ können die Zellen ein Genprodukt wie rekombinante Proteine exprimieren (beispielsweise bei einer Proteinüberexpression). Klonierungen spielen eine Rolle
- in der Molekularbiologie und Biochemie
- zur Untersuchung von Proteinen und deren Funktionen per Expressionsklonierung
- zur Veränderung der Eigenschaften von Proteinen im Zuge eines Protein-Engineerings
- bei der Untersuchung von regulatorischen Elementen
- bei der Generierung von Sonden und anderen molekularbiologischen Werkzeugen (z. B. antisense-Konstrukte oder CRISPR/Cas-Werkzeuge)
- in der Biotechnologie
- für therapeutische Zwecke (z. B. Insulin)
- für eine Verwendung in der Lebensmitteltechnologie (z. B. Lab-Ferment)
- zur Generierung gentechnisch veränderter Organismen in der Landwirtschaft (z. B. Flavr-Savr-Tomate)
Für die Vervielfältigung der Klonierungsprodukte werden verschiedene Organismen als Wirt genutzt. Bekannte Beispiele sind Bakterienzellen wie das Bakterium Escherichia coli, einzellige Algen oder Pilze. Die Wirtszellen vermehren sich dabei durch Zellteilung, wobei auch identische Kopien der zu klonierenden Ziel-DNA hergestellt werden. Das Resultat ist eine Population von Zellen, die alle einen Klon des gewünschten DNA-Fragments enthalten. Aus dieser Population wird zur weiteren Verwendung ein geeigneter Klon isoliert.
Die klonierte DNA kann dazu verwendet werden, ein oder mehrere Gene auf fremde Organismen zu übertragen, um so Stoffwechselprozesse zu verbessern oder Resistenzen zu verleihen (Genmanipulation bei Tieren und Pflanzen). Durch eine funktionelle Klonierung werden ähnliche DNA-Sequenzen identifiziert, durch eine positionelle Klonierung werden benachbarte DNA-Sequenzen identifiziert.
Verfahren
Beim Klonieren werden sogenannte Vektoren („Genfähren“) verwendet. Diese dienen als Transportmittel zur Übertragung einer bestimmten DNA-Sequenz (genannt Transgen oder engl. insert) in eine Empfängerzelle und dessen Vervielfältigung. Um diese Vektoren für Fremd-DNA aufnahmefähig zu machen, gibt es verschiedene Verfahren:
Restriktion und Ligation
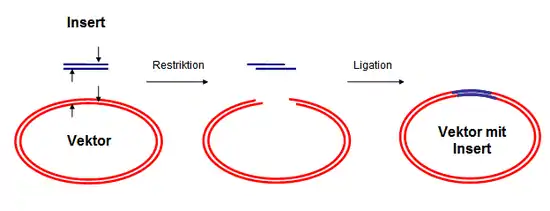
Bei der Plasmidklonierung wird ein Plasmid (beispielsweise pUC19) im Zuge eines Restriktionsverdaus mit Hilfe von speziellen Restriktionsenzymen versetzt geschnitten, so dass überhängende Enden (engl. sticky ends, klebrige Enden) entstehen. Bei der zu inserierenden DNA handelt es sich um ein Fragment, das mit denselben Enzymen aus einem anderen Vektor isoliert wurde, um synthetische DNA mit den passenden Überhängen oder um mittels Polymerase-Kettenreaktion (PCR) aus genomischer DNA oder cDNA amplifizierte DNA. Vor der Inserierung der PCR-Produkte in den Vektor werden diese mit denselben Enzymen geschnitten, so dass komplementäre Enden an Vektor- und Ziel-DNA entstehen. Die zueinander kompatiblen, überhängenden Enden von Vektor- und Ziel-DNA finden sich und hybridisieren miteinander. In der nachfolgenden Ligation, die durch eine DNA-Ligase (z. B. T4-DNA-Ligase) katalysiert wird, werden die Enden der Einzelstränge miteinander kovalent verbunden. Um den Hintergrund an Klonen ohne Inserts zu senken, wird der Vektor zur Entfernung der 5'-Phosphatgruppe vor der Ligation häufig mit Phosphatase, z. B. Calf-Intestine-Phosphatase (CIP), behandelt. Dies erlaubt auch die effiziente Klonierung in Vektoren, die nur mit einem Restriktionsenzym behandelt wurden.
Nach der anschließenden Transformation werden rekombinante Bakterien durch Plattieren auf Agarplatten mit einem geeigneten Antibiotikum selektioniert. Wenn die Vektoren dies erlauben, werden mittels Blau-Weiß-Screening Kolonien ausgemustert, die kein Insert enthalten. Auch damit ist noch nicht gesichert, dass alle anderen Kolonien das gewünschte Insert enthalten. Daher wird die DNA einzelner Kolonien mittels Restriktionsverdau oder Kolonie-PCR charakterisiert. Wenn das Ergebnis nicht eindeutig ist oder das Insert durch DNA-Amplifikation erzeugt wurde, schließt sich eine DNA-Sequenzierung an. Hinterlässt eines der Enzyme, mit dem der Vektor geschnitten wird, ein glattes Ende (engl. blunt end), wird das PCR-Fragment mit nur einem Enzym geschnitten.
PCR-Klonierung
Bei dieser Variante wird die einzufügende DNA-Sequenz in einer PCR mit Primern vervielfältigt, die jeweils eine mit dem Vektor überlappende Sequenz am 5'-Ende enthalten. Anschließend wird das gereinigte PCR-Produkt im Zuge einer Ligation-During-Amplification (einer PCR-verwandten Mutagenese-Reaktion) als Megaprimer verwendet, um das Plasmid in vitro zu synthetisieren. Restriktionsenzyme und DNA-Ligase werden hierbei nicht verwendet. Nach einem Abbau der Ausgangsplasmide werden die neu erzeugten Plasmide zur Vermehrung in Bakterien transformiert. Die Ligation erfolgt nach der Transformation in vivo. Eine Variante der PCR-Klonierung ist das circular polymerase extension cloning (CPEC).[1]
TA-Klonierung
Die TA-Plasmide sind ebenfalls linearisierte Plasmide, die jedoch als letztes Nukleotid ein Thymidinnukleotid als 3'-Überhang besitzen, das als Sticky End wirkt und eine Restriktion der einzufügenden DNA und des Plasmids erübrigt.[2][3] Das überhängende Thyminnukleotid wird zuvor durch eine Desoxyribonukleotidyltransferase (engl. terminal deoxynucleotide transferase) unter Verwendung von Didesoxy-Thyminnukleotiden angefügt.[4] Die einzufügende DNA-Sequenz muss hierbei mit einer thermostabilen DNA-Polymerase vom Typ A (bakteriellen Ursprungs) erzeugt werden, da Typ-B-Polymerasen keinen Überhang erzeugen. Nach einer Ligation wird das Plasmid zur Vermehrung in Bakterien transformiert. Die TA-Klonierung vermeidet eine Verwendung von Restriktionsenzymen, erlaubt aber keine gezielte Orientierung des Inserts im Vektor, wodurch nur in etwa 50 % der transgenen Vektoren eine Genexpression stattfinden kann. Daher werden TA-Vektoren häufig in Kombination mit einem Blau-Weiß-Screening als reine Klonierungsvektoren eingesetzt.
TOPO-Klonierung
TOPO-Plasmide werden durch Restriktionsverdau linearisiert und anschließend kovalent mit einer Topoisomerase aus dem Vacciniavirus (ein Pockenvirus) gekoppelt,[5] die die Ligation eines PCR-Produkts bewirkt und daher keine Ligase mehr erfordert.[6] Die kovalente Bindung der Topoisomerase erfolgt selbsttätig an eine DNA-Sequenz am 3'-Phosphatendende der Sequenz 5´-(C/T)CCTT-3'.[7][8] TOPO-Klonierungsvektoren gibt es als TA-Vektoren und als Blunt-end-Vektoren. Anschließend wird das Plasmid zur Vermehrung in Bakterien transformiert. Da die Orientierung des Inserts im Vektor zufallsgemäß ist, werden diese Vektoren als reine Klonierungsvektoren eingesetzt. Wird schon im ersten Schritt die Generierung eines Expressionsklons angestrebt, werden unidirektionale TOPO-Vektoren eingesetzt. Häufig werden auch unidirektionale Entry-Vektoren für das Gateway-System genutzt. TOPO und Gateway sind eingetragene Warenzeichen der Firma Thermo Fisher Scientific.
Isothermal Assembly
Durch Isothermal Assembly werden durch PCR oder künstliche Gensynthese hergestellte DNA-Fragmente, ohne dass diese zuvor mit Restriktionsenzymen behandelt werden müssen, in einen linearisierten Vektor eingefügt. Oftmals wird das Gibson Assembly eingesetzt, das von Daniel G. Gibson im J. Craig Venter Institute entwickelt wurde.[9] Es erlaubt in einem einzigen Schritt den nahtlosen Einbau (engl. Seamless Cloning) eines oder mehrerer Fragmente in einen Vektor. Voraussetzung ist, dass die zu ligierenden Moleküle Sequenzüberlappungen von etwa 20 Nukleotiden aufweisen. Durch Inkubation mit dNTPs und einem Cocktail aus drei Enzymen, einer Exonuklease (T5-Exonuklease), die die Moleküle vom 5'-Ende her kürzt und damit die Hybridisierung der Moleküle erlaubt, einer DNA-Polymerase (Phusion DNA-Polymerase), die entstandenen Lücken schließt, und einer thermostabilen DNA-Ligase (Taq DNA-Ligase), die einen Ringschluss bewirkt, entstehen Plasmide, die direkt transformiert werden können.[10] Gibson Assembly ist ein eingetragenes Warenzeichen der Firma New England Biolabs.
LIC und SLIC
Bei einer weiteren Klonierungsvariante (engl. Ligation-independent Cloning, LIC) wird das PCR-Produkt mit einer LIC-Sequenz versehen. Mittels der 3’ → 5’ Exonuklease-Aktivität der T4-DNA-Polymerase werden in Anwesenheit eines dNTPs komplementäre Überhänge im Vektor und im PCR-Produkt erzeugt. Die Ligation des annealten Produktes erfolgt nach der Transformation in vivo.[11] Eine Weiterentwicklung der Methode ist die SLIC, die sequenz- und ligationsunabhängige Klonierung, die ohne LIC-Sequenz auskommt.[12] Die Exonuklase-Aktivität wird in SLIC-Protokollen durch Zugabe eines dNTPs gestoppt. Die Lücken in den nach dem Mischen der DNA-Fragmente entstehenden ringförmigen Plasmiden werden nach der Transformation in E.-coli-Zellen repariert.
Gateway-Klonierung
Bei einer Gateway-Klonierung werden Sequenzen an das Transgen angefügt,[13] das Erkennungssequenzen für die Ligase attB enthält, die den Einbau des Transgens in einen entry vector katalysiert. Hierbei wird gleichzeitig im entry vector das Selbstmordgen ccdB entfernt, wodurch nur transgene Organismen mit Vektor heranwachsen. Durch einen anschließenden Austausch von Gensegmenten durch eine Excisionase erfolgt ein Gentransfer vom entry vector in einen Zielvektor, der meistens der Expression des Transgens dient und zur späteren Selektion eine andere Antibiotikum-Resistenz besitzt. Als zweiten Selektionsdruck erhält bei diesem Austausch der entry vector vom Zielvektor das ccdB-Selbstmordgen, wodurch fast nur Organismen mit dem Transgen-enthaltenden Zielvektor heranwachsen.
Golden-Gate-Klonierung
Die Golden-Gate-Klonierung, auch als Golden Gate Assembly bekannt, erlaubt mit Hilfe von Typ-IIs-Restriktionsenzymen und T4-DNA-Ligase den simultanen direktionalen In-vitro-Zusammenbau mehrerer DNA-Fragmente.[14][15][16] Die bei dieser Methode eingesetzten Typ-IIs-Restriktionsenzymen, wie BsaI, BsmBI und BbsI, schneiden außerhalb ihrer Erkennungssequenz und können daher nicht-palindromische, vier Basenpaar lange Überhänge generieren, die nach einer Ligation keine Schnittstellen mehr aufweisen. Da die Restriktionsschnittstellen nicht Teil des entstandenen Konstruktes sind, können Restriktionsverdau und Ligation simultan durchgeführt werden. Die Methode wird u. a. für die Herstellung von TALEN für das Genome Editing genutzt.[17]
Rekombination
Bei den Rekombinationsverfahren wie dem Recombineering und dem RMCE-Kassettenaustauschverfahren erfolgt die Restriktion und Ligation nach gemeinsamer Transformation von Vektor und Insert in vivo.
Künstliche Gensynthese
Durch verschiedene Methoden der künstlichen Gensynthese können die Inserts de novo aus Primern synthetisiert werden. Die synthetisierten Doppelstränge enthalten schon die geeigneten Enden für die Ligation in den gewünschten Vektor. Dies können z. B. überhängende Enden für die Ligation in einen mit Restriktionsenzymen geschnittenen Vektor oder Sequenzüberlappungen für das Gibson Assembly sein.
Transformation und Selektion
Im Anschluss folgt die Transformation kompetenter Bakterienzellen (etwa E. coli) mit dem Vektor-Insert-Konstrukt. Das Antibiotikum-Resistenzgen auf dem Vektor erlaubt die Selektion von Bakterienzellen, die das Plasmid ins Zellinnere aufgenommen haben.[18] Hierzu wird der Transformations-Ansatz auf einem Agar-Nährboden (z. B. LB-Medium) ausplattiert, der das entsprechende Antibiotikum (z. B. Ampicillin oder Kanamycin) enthält. So werden nur diejenigen Bakterienzellen selektiert, die ein Plasmid aufgenommen haben. Durch Vermehrung dieser einzelnen Bakterienzellen entstehen Bakterienkolonien. Alle untransformierten und somit nicht gegen das verwendete Antibiotikum resistenten Bakterien sterben. Falls die Vektoren wie z. B. pUC19 dies erlauben, werden mittels Blau-Weiß-Screening Kolonien ausgemustert, die kein Insert enthalten. Zu diesem Zweck werden Platten verwendet, die neben dem Antibiotikum noch X-Gal und IPTG enthalten. Die Entstehung von blauen Kolonien weist auf das Vorhandensein von Bakterienzellen mit unveränderten Vektoren hin, während die ungefärbten Kolonien möglicherweise das gewünschte Insert enthalten. Daher wird die DNA einzelner Kolonien direkt, oder nach Amplifizierung in Flüssigkultur, mittels Restriktionsverdau oder Kolonie-PCR charakterisiert. Wenn das Ergebnis nicht eindeutig ist oder das Insert durch DNA-Amplifikation erzeugt wurde, schließt sich eine DNA-Sequenzierung an. Zur weiteren Vermehrung impft man mit einer solchen Kolonie ein Flüssigmedium an. Aus einem solchen Ansatz kann eine große Menge Plasmid-DNA isoliert (Plasmidpräparation) werden. Die DNA steht dann für weitere Klonierungen oder Transformationen zur Verfügung.
Literatur
- Michael Andrew Quail: DNA Cloning. In: Encyclopedia of Life Sciences. doi:10.1038/npg.els.0005344 PDF 2001
- Nancy Trun & Janine Trempy: DNA Cloning. In: Fundamental Bacterial Genetics. ISBN 0-632-04448-9 PDF 2003
Einzelnachweise
- J. Quan, J. Tian: Circular polymerase extension cloning of complex gene libraries and pathways. In: PloS one. Band 4, Nummer 7, 2009, S. e6441, ISSN 1932-6203. doi:10.1371/journal.pone.0006441. PMID 19649325. PMC 2713398 (freier Volltext).
- T.A. Holton, Graham, M.W: A simple and efficient method for direct cloning of PCR products using ddT-tailed vectors. In: Nucleic Acids Research. 19, Nr. 5, 11. März 1991, S. 1156. doi:10.1093/nar/19.5.1156. PMID 2020554. PMC 333802 (freier Volltext).
- Y. Ichihara, Y. Kurosawa: Construction of new T vectors for direct cloning of PCR products. In: Gene (1993), Band 130(1), S. 153–154. PMID 8344524.
- M. Y. Zhou, C. E. Gomez-Sanchez: Universal TA cloning. In: Curr Issues Mol Biol. (2000), Band 2(1), S. 1–7. PMID 11464915.
- L. Geng, W. Xin, D. W. Huang, G. Feng: A universal cloning vector using vaccinia topoisomerase I. In: Mol Biotechnol. (2006), Band 33(1), S. 23–28. PMID 16691003.
- C. Cheng, S. Shuman: DNA strand transfer catalyzed by vaccinia topoisomerase: ligation of DNAs containing a 3' mononucleotide overhang. In: Nucleic Acids Res. (2000), Band 28(9), S. 1893–1898. PMID 10756188; PMC 103307 (freier Volltext).
- S. G. Morham, S. Shuman: Covalent and noncovalent DNA binding by mutants of vaccinia DNA topoisomerase I. In: J Biol Chem. (1992), Band 267(22), S. 15984–15992. PMID 1322412.
- J. Sekiguchi, S. Shuman: Proteolytic footprinting of vaccinia topoisomerase bound to DNA. In: J Biol Chem. (1995), Band 270(19), S. 11636–11645. PMID 7744804.
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, Smith HO.: Enzymatic assembly of DNA molecules up to several hundred kilobases. In: Nature Methods. 6, Nr. 5, 2009, S. 343–345. doi:10.1038/nmeth.1318. PMID 19363495.
- R. M. Benoit, C. Ostermeier, M. Geiser, J. S. Li, H. Widmer, M. Auer: Seamless Insert-Plasmid Assembly at High Efficiency and Low Cost. In: PloS one. Band 11, Nummer 4, 2016, S. e0153158, doi:10.1371/journal.pone.0153158, PMID 27073895, PMC 4830597 (freier Volltext).
- R. S. Haun, I. M. Serventi, J. Moss: Rapid, reliable ligation-independent cloning of PCR products using modified plasmid vectors. In: BioTechniques (1992), Band 13(4), S. 515–518. PMID 1362067.
- M. Z. Li, S. J. Elledge: Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. In: Nature methods. Band 4, Nummer 3, März 2007, S. 251–256, doi:10.1038/nmeth1010, PMID 17293868.
- D. Esposito, L. A. Garvey, C. S. Chakiath: Gateway cloning for protein expression. In: Methods in molecular biology. Band 498, 2009, S. 31–54, doi:10.1007/978-1-59745-196-3_3, PMID 18988017.
- C. Engler, R. Kandzia, S. Marillonet: A One Pot, One Step, Precision Cloning Method with High Throughput Capability. In: PLOS One Band 3(11), 2008, e3647, PMID 18985154.
- E. Weber, C. Engler, R. Gruetzner, S. Werner, S. Marillonnet: A modular cloning system for standardized assembly of multigene constructs. In: PloS one. Band 6, Nummer 2, 2011, S. e16765, doi:10.1371/journal.pone.0016765, PMID 21364738, PMC 3041749 (freier Volltext).
- C. Engler, R. Kandzia, S. Marillonnet: A one pot, one step, precision cloning method with high throughput capability. In: PloS one. Band 3, Nummer 11, 2008, S. e3647, doi:10.1371/journal.pone.0003647, PMID 18985154, PMC 2574415 (freier Volltext).
- N.E. Sanjana, L. Cong, Y. Zhou, M. M. Cunniff, G. Feng, F. Zhang: A Transcription Activator-Like Effector Toolbox for Genome Engineering. In: Nature Protocols Band 7(1), (2012), S. 171–192. PMID 22222791.
- Ulrich Helmich: Nachweis klonierter Zellen