Arzneimittelzulassung
Eine Arzneimittelzulassung ist eine behördlich erteilte Genehmigung, die erforderlich ist, um ein industriell hergestelltes, verwendungsfertiges Arzneimittel anbieten, vertreiben oder abgeben zu können. Eine Arzneimittelzulassung wird immer nur für eine bestimmte Indikation (d. h. ein Anwendungsgebiet) erteilt. Der Einsatz eines zugelassenen Arzneimittels außerhalb der genehmigten Indikation wird als Off-Label-Anwendung bezeichnet.
Zweck des Verfahrens
Der Zweck eines Zulassungsverfahrens für Stoffe und Zubereitungen als Arzneimittel ist die Risikovorsorge und Abwehr von Gefährdungen der Gesundheit, die durch unsichere oder wirkungslose Arzneimittel entstehen könnten. Im Rahmen des Zulassungsverfahrens werden deshalb vom Pharmaunternehmen eingereichte Unterlagen zur pharmazeutischen Qualität, therapeutischen Wirksamkeit und Unbedenklichkeit des Arzneimittels durch Arzneimittelbehörden überprüft; die Angaben in den Unterlagen werden durch Inspektionen vor Ort kontrolliert.
Wirkungen der Zulassung
Mit der erteilten Zulassung wird bescheinigt, dass das Arzneimittel verkehrsfähig ist und auf den Markt gebracht werden darf, also beispielsweise in Apotheken angeboten werden kann. Für die Ärzte ist die Zulassung ein Nachweis dafür, dass das Arzneimittel in der angegebenen Indikation auf ein positives Nutzen-Risiko-Verhältnis geprüft wurde. Die erteilte Zulassung hat aber noch weitere Wirkungen.
In der Regel ist die Arzneimittelzulassung eine Voraussetzung für die sozialrechtliche Versorgungsfähigkeit. In vielen Ländern gibt es darüber hinaus weitere, von der arzneimittelrechtlichen Zulassung unabhängige, sozialrechtliche Zulassungsverfahren, die eine zusätzliche Voraussetzung für eine Erstattung der Arzneimittelkosten durch die Krankenkassen darstellen. Diese Regelungen sind von Land zu Land verschieden, sie beinhalten oft auch eine verbindliche Preisfestsetzung und können den Marktzugang um etliche Monate verzögern.
Ferner existieren in manchen Ländern besondere Haftungsregelungen für zugelassene Arzneimittel. In Deutschland gibt es eine besondere Gefährdungshaftung auf der Grundlage von § 84 AMG, nach der das Pharmaunternehmen bei Schädigungen durch Arzneimittel, die bestimmungsgemäß angewendet wurden, Schadensersatz zu leisten hat. Dies gilt sowohl bei Herstellungs- als auch bei Entwicklungsfehlern. Der Geschädigte muss, im Gegensatz zur Haftung nach BGB, die Kausalität zwischen der Anwendung und dem entstandenen Schaden nicht nachweisen. Diese Haftung greift nicht beim Off-Label-Use. Österreich hat in seinem Arzneimittelgesetz im Gegensatz zu Deutschland keine besondere Gefährdungshaftung eingeführt.
Eine gültige Zulassung ist in der EU auch für die Erteilung eines ergänzenden Schutzzertifikates gemäß der Verordnung (EWG) Nr. 469/2009 (ersetzt Nr. 1768/92) notwendig. Ein solches Zertifikat kann die Laufzeit eines Patentes auf das Arzneimittel um bis zu 5 Jahre verlängern.
Pflicht zum Zulassungsverfahren
In den verschiedenen Rechtsordnungen gibt es unterschiedliche Bestimmungen dazu, welche Arzneimittel der Pflicht des Zulassungsverfahrens unterliegen. In der Europäischen Union ist nach Artikel 2 der Richtlinie 2001/83/EG das Europäische Arzneimittelrecht auf solche Arzneimittel anzuwenden, die in den Mitgliedstaaten in den Verkehr gebracht werden sollen und die entweder gewerblich zubereitet werden oder bei deren Zubereitung ein industrielles Verfahren zur Anwendung kommt. Das deutsche Arzneimittelrecht bestimmt die Zulassungspflicht für Fertigarzneimittel, das österreichische Recht verwendet analog dazu den Begriff Arzneispezialität. Darunter sind im Voraus hergestellte Arzneimittel zu verstehen, die unter der gleichen Bezeichnung in einer zur Abgabe an den Verbraucher oder Anwender bestimmten Packung abgegeben werden. In der Schweiz gilt die Zulassungspflicht für verwendungsfertige Arzneimittel.
Dabei wird allgemein ein umfassender Arzneimittelbegriff zugrunde gelegt, unter den beispielsweise auch Impfstoffe, viele Blutprodukte und In-vivo-Diagnostika fallen. Dabei gibt es allerdings im Einzelfall Probleme mit der Produktabgrenzung.
Zulassungspflichtige Arzneimittel machen heute den überwiegenden Teil der gebräuchlichen Arzneimittel aus. Es gibt aber eine ganze Reihe von Ausnahmen, die nach wie vor nicht unter die Zulassungspflicht fallen. In der Europäischen Union können homöopathische Arzneimittel und traditionelle pflanzliche Arzneimittel nach einem vereinfachten Genehmigungsverfahren (in Deutschland „Registrierung“ genannt) in den Verkehr gebracht werden, sofern sie die entsprechenden, gesetzlich geregelten Voraussetzungen dafür erfüllen. In diesem vereinfachten Verfahren müssen lediglich Qualität und Unbedenklichkeit nachgewiesen werden; bei registrierten Homöopathika darf dann keine Indikation angegeben werden, bei traditionell verwendeten pflanzlichen Arzneimitteln muss in der Indikationsformulierung auf die traditionelle Verwendung Bezug genommen werden. Analog dazu sieht die Schweiz für Arzneimittel der Komplementärmedizin ein vereinfachtes Zulassungsverfahren vor. Nicht zulassungspflichtig sind in Apotheken hergestellte Rezeptur- und Defekturarzneimittel sowie Prüfpräparate für klinische Studien. Unter bestimmten Bedingungen können (noch) nicht zugelassene Arzneimittel den Patienten im Rahmen des Compassionate Use zur Verfügung gestellt werden.
Harmonisierung
Seit 1990 wurden im Rahmen des International Council for Harmonisation (ICH) wesentliche Arzneimittelprüfleitlinien zum Nachweis der Qualität, Sicherheit und Wirksamkeit sowie Dokumentenformate für die Zulassungsunterlagen harmonisiert. Die ICH wird von Behörden und Industrievertretern aus Europa, den USA und Japan getragen. Ziel der Harmonisierung war es, dass nichtklinische und klinische Studien aus einer Region in den anderen Regionen anerkannt werden, sodass eine mehrfache Durchführung entfällt. Die einzelnen Behörden sind aber in ihrer Beurteilung der Zulassungsanträge unabhängig, somit kommt es immer wieder vor, dass bestimmte Arzneimittel nicht in allen Regionen zugelassen sind.
Kriterien der Zulassung
Das wichtigste Zulassungskriterium ist die Abwägung des Nutzen-Risiken-Verhältnisses; nur wenn der Nutzen des Arzneimittels die Risiken überwiegt, ist eine Zulassung gerechtfertigt. Wird das Nutzen-Risiken-Verhältnis des Arzneimittels nach erteilter Zulassung ungünstig, muss das Arzneimittel vom Markt genommen werden. Eine Arzneimittelzulassung wird immer nur für ein bestimmtes Anwendungsgebiet, eine bestimmte Indikation erteilt. Die Anwendung eines zugelassenen Arzneimittels außerhalb der genehmigten Indikation wird als Off-Label-Use bezeichnet.
Prüfung der Zulassungsunterlagen
Der Schwerpunkt des Zulassungsverfahrens ist die Sichtung der vom Pharmaunternehmen vorgelegten Unterlagen, die die Qualität, Wirksamkeit und Unbedenklichkeit belegen sollen. Dabei wird von der Arzneimittelbehörde auch geprüft, ob in den verschiedenen Phasen der Arzneimittelstudie nachgewiesen wurde, dass die Herstellung, Qualitätskontrolle, nichtklinische Prüfung und klinische Prüfung (Phase III) nach den vorgeschriebenen Arzneimittelprüfrichtlinien und den empfohlenen internationalen Leitlinien durchgeführt wurden und dem Stand der Wissenschaft entsprechen. Die Arzneimittelbehörde beschränkt sich aber nicht auf die Prüfung der vom Antragsteller eingereichten Unterlagen, sondern kontrolliert durch Inspektionen vor Ort, ob die Studien nach den Regeln der Guten Arbeitspraxis (GxP) durchgeführt wurden.
Erforderliche Zulassungsunterlagen
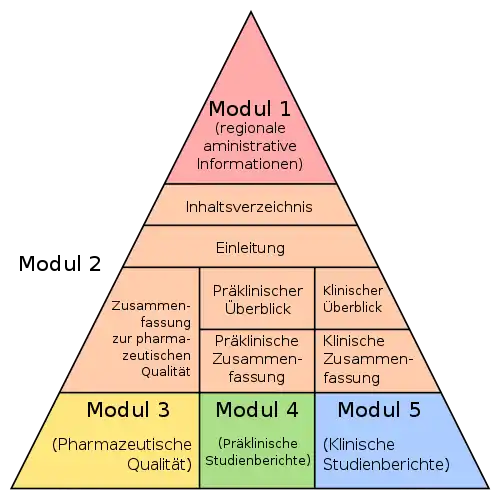
Das Pharmaunternehmen muss mit dem Zulassungsantrag ein umfangreiches Dossier zum Arzneimittel in einem definierten Format, dem Common-Technical-Document-Format (CTD) einreichen. Das CTD-Dossier enthält in fünf Modulen alle Ergebnisse zur Herstellung, Forschung und Entwicklung für das betreffende Arzneimittel. Im Einzelnen enthält das Dossier im CTD Modul 1 regional spezifische Informationen und im CTD Modul 2 einen Überblick und Zusammenfassungen der folgenden Module. CTD Modul 3 enthält einen Qualitätsteil, der beschreibt, wie das Arzneimittel in hinreichender pharmazeutischer Qualität hergestellt werden kann und wie dieses analysiert und nachgewiesen wird, CTD Modul 4 die vorgeschriebenen nichtklinischen pharmakologischen und toxikologischen Studien; CTD Modul 5 umfasst sämtliche Daten aus den klinischen Studien.
Das CTD-Format wurde in der ICH entwickelt. Ziel bei der Einführung des CTD war es, dass in verschiedenen Regionen weitgehend identische Dossiers eingereicht werden können. Inzwischen ist das Format in Europa, Nordamerika und Japan vorgeschrieben. Viele Antragsteller reichen ihre Dossiers inzwischen elektronisch in Form eines eCTD ein.
Zum fortdauernden Nachweis der Arzneimittelsicherheit müssen Pharmaunternehmen zahlreiche rechtliche Anforderungen[1] erfüllen, u. a. muss eine sachkundige Person ernannt sein, es muss einen Risiko-Management-Plan geben und auch die jederzeitige Erreichbarkeit verantwortlicher Beauftragter muss sichergestellt sein.
Pharmazeutische Qualität
Die pharmazeutische Qualität eines Arzneimittels ist die Zusammensetzung des Arzneimittels nach Art und Menge der Bestandteile. Das deutsche Arzneimittelgesetz definiert Qualität als die Beschaffenheit eines Arzneimittels, die nach Identität, Gehalt, Reinheit, sonstigen chemischen, physikalischen, biologischen Eigenschaften oder durch das Herstellungsverfahren bestimmt wird.[2]
Für die Zulassung ist es erforderlich, dass das Arzneimittel eine nach anerkannten pharmazeutischen Regeln angemessene Qualität aufweist. Diese Regeln sind unter anderem in Arzneibuch-Monografien niedergelegt. Die vorzulegenden Unterlagen beschränken sich nicht nur auf die Zusammensetzung des Arzneimittels. Das gesamte Herstellungsverfahren und die Kontrollen der Ausgangsstoffe, Verpackungsmaterialien, Zwischenprodukte und Fertigprodukte sowie durchgeführte Haltbarkeitsstudien sind ausführlich zu dokumentieren.
Noch vor der Zulassung eines Arzneimittels muss der Hersteller eine Herstellungserlaubnis erlangen. Die Herstellung muss nach den Regeln der Good Manufacturing Practice erfolgen; dies wird durch Behörden vor Ort inspiziert. Bei Bedarf können Proben des Arzneimittels in amtlichen Arzneimitteluntersuchungsstellen auf seine pharmazeutische Qualität geprüft werden.
Im Amerikanischen ist die Bezeichnung Chemistry, Manufacturing and Controls (CMC) gebräuchlich.[3]
Wirksamkeit
Die Wirksamkeit eines Arzneimittels ist die Summe der erwünschten Wirkungen im vorgesehenen Anwendungsgebiet.[4] Die erwünschten Wirkungen können eine Heilung, Besserung oder Verhinderung der Krankheit sein. Zur Zulassung wird eine angemessene Wirksamkeit des Arzneimittels in der angestrebten Indikation gefordert. Darunter ist keine Erfolgsgarantie bei jedem Patienten zu verstehen, sondern eine Wahrscheinlichkeit, dass mit dem Arzneimittel therapeutische oder präventive Ergebnisse erzielt werden können. Es gibt mehrere zulässige Verfahren für die Bestimmung der Wirksamkeit, die sich in ihrem Evidenzgrad unterscheiden. Nach Möglichkeit sind randomisierte kontrollierte Studien zu klinischen Endpunkten wie Morbidität, Hospitalisierung und Mortalität durchzuführen.[5] Es gibt aber seitens der Zulassungsstellen anerkannte Gründe dafür, dass die Wirksamkeit auf Grund des Studien-Designs und/oder der erfassten Parameter lediglich mit geringer Evidenz wahrscheinlich gemacht wird, beispielsweise durch epidemiologische Vergleiche oder das Erfassen von Surrogatmarkern.[6][7][8] Nach einem Urteil des deutschen Bundesverwaltungsgerichtes meint der Begriff der therapeutischen Wirksamkeit die „Ursächlichkeit der Anwendung des Arzneimittels für den Heilungserfolg.“[9] Die Studien müssen nach den Regeln der Good Clinical Practice durchgeführt worden sein; die Behörden überprüfen dies durch Inspektionen vor Ort.
Bei Studien zum Wirksamkeitsnachweis ist es wichtig, dass die beobachteten Gruppen von Patienten aus Personen bestehen, die sich in ihren Eigenschaften gleichen und den Patienten im klinischen Alltag entsprechen. Die Gruppen dürfen sich beispielsweise in Alter, Geschlechterverteilung und Schwere der Krankheit nicht unterscheiden. Anderenfalls wäre mit systematischen Fehlern (Bias) und einer fraglichen Alltagsrelevanz zu rechnen. Kann dieses Problem nicht durch eine Randomisierung gelöst werden, bietet sich das Verfahren des Propensity score matching an. Hierbei werden die Probanden der Fall- und Kontrollgruppe nach Ihren Eigenschaften quasi paarweise geordnet und somit gleichmäßig verteilt.[10]
Unbedenklichkeit
Unter dem Aspekt der Unbedenklichkeit wird in erster Linie die mögliche Schädlichkeit eines Arzneimittels beurteilt. Der Begriff Unbedenklichkeit ist seit langem im deutschen Arzneimittelrecht gebräuchlich; international wird meist von Sicherheit gesprochen. Bedenklich sind nach dem deutschen Arzneimittelgesetz Arzneimittel, bei denen nach dem jeweiligen Stand der wissenschaftlichen Erkenntnisse der begründete Verdacht besteht, dass sie bei bestimmungsgemäßem Gebrauch schädliche Wirkungen haben, die über ein nach den Erkenntnissen der medizinischen Wissenschaft vertretbares Maß hinausgehen.[11]
Es gibt kein absolutes Maß dafür, welche Nebenwirkungen hinnehmbar sind; die Bedenklichkeit ist nur in Abwägung zur Schwere der zu behandelnden Krankheit zu beurteilen. Bei der Beurteilung der Unbedenklichkeit eines Arzneimittels ist somit auch die Wirksamkeit heranzuziehen. Unbedenklichkeit bedeutet nicht Unschädlichkeit. Von dem Pharmakologen Gustav Kuschinsky stammt der Spruch, dass für Arzneimittel, von denen behauptet werde, dass es keine Nebenwirkungen habe, der Verdacht bestehe auch keine Hauptwirkung zu besitzen.
Die Unbedenklichkeit des Arzneimittels muss in nichtklinischen und klinischen Studien nachgewiesen werden. Die nichtklinische Prüfung enthält eine umfassende Toxizitätsbestimmung, die in geeigneten in vitro- und Tierversuchen durchzuführen ist. In klinischen Studien werden alle Nebenwirkungen und schwerwiegenden unerwünschten Ereignisse bei den Teilnehmern sorgfältig dokumentiert und ausgewertet.
Problematisch ist, dass die Nebenwirkungen in klinischen Studien nur an einer vergleichsweise geringen Zahl von Patienten dokumentiert werden können; seltene und sehr seltene Nebenwirkungen sind so prinzipiell nicht feststellbar. Das Gleiche gilt aufgrund der begrenzten Laufzeit der Studien für Langzeitfolgen. Es kann sich also zum Zeitpunkt der Zulassung nur um eine vorläufige Beurteilung handeln. Ein vollständiges Sicherheitsprofil kann sich erst bei breiter Anwendung und sorgfältiger Anwendungsüberwachung durch die Pharmakovigilanz ergeben.
Nutzen-Risiko-Verhältnis
Das Nutzen-Risiko-Verhältnis eines Arzneimittels ist das Verhältnis der Wirksamkeit bei der Behandlung einerseits und allen möglichen Risiken im Zusammenhang mit der Qualität, Sicherheit oder Wirksamkeit des Arzneimittels andererseits. Dieses Verhältnis ist von zentraler Bedeutung für die Entscheidung über die Zulassung des Arzneimittels.[12] Dabei reicht der Begriff Nutzen-Risiko-Verhältnis weiter als der Begriff der Unbedenklichkeit, da nicht nur die Risiken für den Patienten, sondern auch für die öffentliche Gesundheit und die Umwelt zu berücksichtigen sind.
Für dieses Verhältnis gibt es keinen allgemein anerkannten Maßstab; dies erfordert immer einen Einzelentscheid. Ähnlich wie bei der Unbedenklichkeit kann die Beurteilung des Nutzen-Risiko-Verhältnisses zum Zeitpunkt der Zulassung nur vorläufig sein. Die mangelnde Generalisierbarkeit des aus den Zulassungsstudien abgeleiteten Nutzen-Risiko-Verhältnisses wurde beispielsweise für nichtsteroidale Antirheumatika exemplarisch gezeigt.[13]
Deshalb soll das Nutzen-Risiko-Verhältnis nach der Zulassung in der Pharmakovigilanz laufend überprüft werden; wird das Verhältnis ungünstig, ist das Mittel vom Markt zu nehmen. Obwohl der Begriff des Nutzen-Risiko-Verhältnisses in der Arzneimittelbewertung seit vielen Jahren gebräuchlich ist, wurde er erst 2004 mit der Reform des EU-Arzneimittelrechts in verschiedene Artikel der EG-Richtlinien und Verordnungen eingefügt.
Für die arzneimittelrechtliche Zulassung ist der Nachweis einer den Risiken überlegenen Wirksamkeit hinreichend, eine Überlegenheit gegenüber anderen Arzneimitteln wird nicht gefordert. In jüngster Zeit wurden in vielen Ländern weitere Beurteilungsverfahren zur Nutzenbewertung oder zur Kosten-Nutzen-Bewertung von Arzneimitteln eingeführt, beispielsweise in Deutschland durch das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) oder im Vereinigten Königreich durch das National Institute for Health and Clinical Excellence (NICE). Diese Beurteilungsverfahren sind nicht Teil der Arzneimittelzulassung.[14] Sie dienen der Beurteilung der Erstattungsfähigkeit von Arzneimitteln durch Krankenkassen.
Zusammenfassung der Merkmale des Arzneimittels

Im Verlauf des Zulassungsverfahrens werden die wesentlichen, durch Studienergebnisse belegten und zwischen Antragsteller und Zulassungsbehörde im Wortlaut vereinbarten Informationen zum Arzneimittel in der Zusammenfassung der Merkmale des Arzneimittels zusammengefasst. Dieses wichtige Dokument enthält alle wesentlichen Informationen wie Indikation, Kontraindikation, Dosierung, Wechselwirkungen und Nebenwirkungen einschließlich der Bewertung und Abwägung. Diese Zusammenfassung kann auch nach der Zulassung nur mit Genehmigung der Behörde geändert werden.
Zulassungsverfahren
Zulassungsverfahren werden durch nationale Regelungen und durch internationale Übereinkommen geregelt. Darüber hinaus wird der eine Zulassung begründende medizinische Hintergrund durch die Entwicklungen von der Konsens-basierten Medizin zur Evidenz-basierten Medizin fortlaufend weiter entwickelt.
Europäische Union
Im Zuge der Verwirklichung des Europäischen Binnenmarktes wurden 1995 vereinheitlichte Verfahren zur EU-Zulassung eingeführt, so dass nicht mehr in jedem EU-Land unterschiedliche bürokratische Hürden überwunden werden müssen. Dazu wurde der Rechtsrahmen mit der Richtlinie 2001/83/EG zur Schaffung eines Gemeinschaftskodexes für Humanarzneimittel sowie der Richtlinie 2001/82/EG zur Schaffung eines Gemeinschaftskodexes für Tierarzneimittel (ab 2022 abgelöst durch die Verordnung (EU) 2019/6 über Tierarzneimittel) in der jeweils aktuellen Fassung EU-weit harmonisiert. Es wurden verschiedene Verfahren der EU-Zulassung entwickelt, die teils auf der Koordinierung dezentraler Institutionen, teils auf Zentralisierung beruhen. Die nationalen Behörden üben aber nach wie vor in jedem Fall Schlüsselfunktionen aus.
Zentralisiertes Verfahren
Das wichtigste Verfahren für innovative Arzneimittel ist das Zentralisierte Verfahren[15] auf der Grundlage der Verordnung (EG) Nr. 726/2004. Dieses Verfahren ist für eine Reihe von Arzneimitteln zwingend vorgeschrieben. Dazu gehören Arzneimittel für neuartige Therapien und monoklonale Antikörper sowie Humanarzneimittel mit neuen Wirkstoffen zur Behandlung von Aids, Diabetes mellitus, Krebs, neurodegenerativen Erkrankungen, Autoimmunerkrankungen und anderen Immunschwächen und Viruserkrankungen. Auch Orphan-Arzneimittel zur Behandlung von seltenen Krankheiten sowie Tierarzneimittel zur Leistungssteigerung fallen zwingend unter das Verfahren. Für eine Reihe weiterer Arzneimittel ist der Zugang zu diesem Verfahren fakultativ möglich.
Im zentralisierten Verfahren sind die wissenschaftliche Beurteilung und die Zulassungsentscheidung institutionell getrennt. Der Zulassungsantrag muss bei der Europäischen Arzneimittelagentur (EMA) eingereicht werden. Das Beurteilungsverfahren für den Zulassungsantrag wird von den wissenschaftlichen Ausschüssen – bei Humanarzneimitteln der Ausschuss für Humanarzneimittel, bei Tierarzneimitteln der Ausschuss für Tierarzneimittel – der Europäischen Arzneimittelagentur durchgeführt; in diese Ausschüsse werden von den Mitgliedsstaaten hochrangige Vertreter der nationalen Arzneimittelbehörden entsandt. Ein aus dem zuständigen wissenschaftlichen Ausschuss der Agentur ausgewählter Berichterstatter und ein Mitberichterstatter erstellen mit Experten aus den nationalen Arzneimittelbehörden einen Beurteilungsbericht für das Arzneimittel, der nach spätestens 210 Tagen vom zuständigen wissenschaftlichen Ausschuss der Europäischen Arzneimittelagentur verabschiedet wird. Auf der Grundlage eines positiven Gutachtens (positive opinion) erteilt die Europäische Kommission nach Konsultierung der Mitgliedsstaaten im Ständigen Ausschuss innerhalb von 67 Tagen die Zulassung für die gesamte Europäische Union. Im zentralisierten Verfahren arbeiten somit nationale und europäische Institutionen eng zusammen. Die erteilte EU-Zulassung wird regelmäßig im Europäischen Wirtschaftsraum (EWR) übernommen.
Ob ein Arzneimittel zentral zugelassen wurde, kann der Verbraucher anhand der Zulassungsnummer erkennen; die Nummer beginnt dann mit der Kennung „EU“. Zum Beispiel lautet eine der Zulassungsnummern für Humira EU/1/03/256/007. Die zweite Stelle gibt an, ob es sich um ein Tier- oder Humanarzneimittel handelt (1= human, 2= veterinär). Die dritte Stelle gibt das Jahr der ersten Zulassung an, hier 2003.
Von 1995 bis September 2007 wurden im zentralisierten Verfahren zirka 400 Humanarzneimittel und 75 Tierarzneimittel zugelassen. Für jedes neu zugelassene Arzneimittel wird ein ausführlicher europäischer öffentlicher Beurteilungsbericht (EPAR) veröffentlicht.
Dezentrale Verfahren (MRP und DCP)
Bei dem Verfahren der gegenseitigen Anerkennung (MRP) und dem ähnlichen Dezentralisierten Verfahren (DCP) wird ein Zulassungsantrag von der Behörde eines Mitgliedstaates (Referenzmitgliedstaat) geprüft und ein Beurteilungsbericht erstellt.[16] In einem koordinierten Prozess erkennen dann die Behörden der anderen betroffenen Mitgliedstaaten diese Beurteilung an. Der Antragsteller kann dabei auswählen, für welche Mitgliedstaaten der EU und des EWR er die Zulassung beantragen will.
Beim Verfahren der gegenseitigen Anerkennung wird erst in einem Land der Wahl eine nationale Zulassung beantragt und erteilt, bevor dann in den anderen Staaten identische Anträge eingereicht werden und das Anerkennungsverfahren in Gang gesetzt wird. Im 2005 eingeführten Dezentralisierten Verfahren darf noch keine nationale Zulassung in der EU vorliegen; hier werden identische Anträge gleichzeitig in allen Staaten eingereicht und ein Staat als Referenzmitgliedstaat ausgewählt.
Wenn einzelne Mitgliedstaaten die Beurteilung des Referenzmitgliedstaates wegen einer schwerwiegenden Gefahr für die öffentliche Gesundheit ablehnen, müssen alle beteiligten Staaten sich in einer Koordinierungsgruppe bemühen, eine Einigung über die zu treffenden Maßnahmen zu erzielen. Können sich die Behörden in der Koordinierungsgruppe nicht einigen, dann kommt es zu einem Schiedsverfahren im wissenschaftlichen Ausschuss der Europäischen Arzneimittelagentur. Auf der Grundlage der Beurteilung durch den wissenschaftlichen Ausschuss fällt die Europäische Kommission im Schiedsverfahren nach Konsultierung der Mitgliedstaaten im Ständigen Ausschuss eine endgültige Entscheidung.
Die Zulassung wird in den nicht zentralisierten Verfahren in jedem Fall von den nationalen Behörden erteilt.
Jährlich werden mehrere hundert nicht zentralisierte Verfahren durchgeführt; darunter sind ein großer Teil Generika.[17]
Nationale Verfahren
Bis 1995 waren nationale Verfahren die einzige Möglichkeit, ein Arzneimittel in der EU zuzulassen. Diese nationalen Verfahren haben durch die europäischen Verfahren viel an Bedeutung verloren. Es sind aber viele Arzneimittel auf dem Markt, die über solche Verfahren zugelassen wurden. Heute ist eine rein nationale Zulassung nur in einem Mitgliedsland möglich; nationale Zulassungsanträge in mehr als einem Mitgliedsland sind nicht mehr zulässig. Üblicherweise ist heute solch eine nationale Zulassung der Einstieg in das Verfahren der gegenseitigen Anerkennung. In Deutschland sind das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) für „normale“ Arzneimittel, das Paul-Ehrlich-Institut für Blutprodukte und Impfstoffe, das Friedrich-Loeffler-Institut für immunologische Tierarzneimittel gegen exotische Tierseuchen sowie das Bundesamt für Verbraucherschutz und Lebensmittelsicherheit für Tierarzneimittel zuständig. In Österreich werden Arzneimittel durch die Österreichische Agentur für Gesundheit und Ernährungssicherheit Bereich Medizinmarktaufsicht (vormals AGES PharmMed) zugelassen und überwacht.
Schweiz
Da die Schweiz weder Mitglied der Europäischen Union noch des Europäischen Wirtschaftsraumes ist, führt die Schweiz sämtliche Arzneimittelzulassungen autonom durch; es können aber grundsätzlich Zulassungen aus Ländern mit vergleichbarer Arzneimittelkontrolle berücksichtigt werden. Die Rechtsgrundlage ist das Heilmittelgesetz, das neben Arzneimitteln auch Medizinprodukte regelt. Details zur Zulassung finden sich in der Arzneimittel-Zulassungsverordnung. Die für die Zulassung und Arzneimittelüberwachung zuständige Einrichtung ist die Swissmedic. Die interne Bearbeitungsfrist für Zulassungsgesuche liegt bei 200 Tagen, für Gesuche im beschleunigten Verfahren bei 130 Tagen. Für bestimmte Arzneimittel, darunter solche mit bekannten Wirkstoffen, Arzneimitteln der Komplementärmedizin, Spitalpräparate und Arzneimittel gegen seltene oder lebensbedrohende Krankheiten gibt es ein vereinfachtes Zulassungsverfahren.
USA
In den USA ist die Arzneimittelzulassung ein Prozess, der deutlich früher einsetzt als in Europa.[18]
Im Prinzip beginnt das Verfahren in den USA bereits mit dem Antrag auf Genehmigung der ersten klinischen Studie (Investigational New Drug application, IND). Dort werden im Gegensatz zum Genehmigungsverfahren für klinische Studien in Europa nicht nur Zusammenfassungen, sondern vollständige Studienberichte eingereicht, die dann im Verlauf der klinischen Entwicklung in einer rollierenden Einreichung laufend ergänzt werden können (rolling submission). Dann liegen beim eigentlichen Zulassungsantrag, der New Drug Application, NDA, viele Unterlagen bereits bewertet vor. Sobald die NDA von der Food and Drug Administration (FDA) als vollständig und den formalen Anforderungen entsprechend akzeptiert ist, wird der Antrag innerhalb einer festgesetzten Frist von der FDA geprüft. Nach Anhörung einer Expertenkommission und des Pharmaunternehmens entscheidet die FDA, ob das Arzneimittel zugelassen wird, ob der Antrag zulässig ist (approvable), was bedeutet, dass die FDA sich bereiterklärt, das Arzneimittel unter bestimmten, vom Antragsteller zu erfüllenden Bedingungen zuzulassen, oder ob der Antrag abgelehnt wird (not approvable).
Nur 10 % aller Arzneimittelentwicklungen, bei denen mit klinischen Studien am Menschen begonnenen wurde, erhalten die Zulassung durch die FDA.[19]
Unter bestimmten Voraussetzungen genügt nach der Animal Efficacy Rule ein Wirksamkeitsnachweis im Tierversuch.
Internationale Anerkennung von Zulassungsentscheidungen
Auch wenn viele Kriterien der Arzneimittelzulassung in vielen Ländern harmonisiert wurden und von den Pharmaunternehmen oft praktisch identische Unterlagen eingereicht werden, kommen die Behörden im Einzelfall doch zu gegensätzlichen Entscheidungen. Das liegt daran, dass die Behörden bei der Zulassung einen großen Beurteilungsspielraum haben und wesentliche Aussagen zum Arzneimittel von Wahrscheinlichkeitsangaben getragen werden.
In der Europäischen Union hat es drei Jahrzehnte gedauert, bis sich zwischen den nationalen Behörden genügend gegenseitiges Verständnis entwickelt hatte, um den Erfolg der EU-Zulassungsverfahren zu ermöglichen.
Von diesem Punkt sind die europäischen Behörden einerseits und die FDA andererseits weit entfernt. Beispielsweise wurde 2006 in der Europäischen Union ein Rimonabant-haltiges Arzneimittel zugelassen, während die FDA 2007 einen Zulassungsantrag für dasselbe Arzneimittel wegen Sicherheitsbedenken abwies. Umgekehrt wurde das Krebsarzneimittel Mylotarg mit dem monoklonalen Antikörper Gemtuzumab-Ozogamicin schon 2000 in den USA zugelassen, während der Zulassungsantrag in der EU 2008 abgelehnt wurde.
Verschiedene Bestrebungen, Arzneimittelzulassungen international gegenseitig anzuerkennen, führten dementsprechend bisher nur zu Teilerfolgen. Mutual Recognition Agreements zur gegenseitigen Anerkennung von Inspektionen zur Good Manufacturing Practice wurden zwischen der Europäischen Union, Australien, Japan, Kanada, Neuseeland, der Schweiz und den USA geschlossen. Das Abkommen mit den USA wurde bisher nicht umgesetzt, das mit Japan nur teilweise.[20]
Besondere Verfahren und Kategorien
Sonderwege für schnelleren Zugang
Die Arzneimittelentwicklung und Zulassung sind ein langwieriger, mehrjähriger Prozess. Um den Zugang zu innovativen, möglicherweise lebensrettenden Arzneimitteln nicht unnötig zu verzögern, wurden in der Europäischen Union und den USA besondere Verfahren eingeführt, die in Ausnahmefällen die Zulassung beschleunigen sollen.
Das beschleunigte Beurteilungsverfahren (accelerated assessment) im zentralisierten Verfahren der EU[21] und das Priority Review-Verfahren bei der FDA haben deutlich verkürzte Bearbeitungszeiten gegenüber den regulären Verfahren. Dies kann die Zulassung um mehrere Monate beschleunigen; in der EU reduziert sich die Bearbeitungszeit im wissenschaftlichen Ausschuss von 210 auf 150 Tage. Die Behörden prüfen im Einzelfall, ob ein Antrag in diesem Verfahren bearbeitet wird. In den USA wurden allein bis 2006 jährlich zirka 10 bis 15 Arzneimittel im Priority Review bearbeitet und zugelassen,[22][23] in der EU wurde mit Eculizumab im Sommer 2007 das erste Arzneimittel im beschleunigten Verfahren zugelassen.[24]
Die bedingte Zulassung (conditional marketing authorisation) im zentralisierten Verfahren der EU[25] auf der Grundlage der Verordnung 507/2006/EG ermöglicht es im Einzelfall, insbesondere bei lebensbedrohenden Krankheiten, ein Arzneimittel noch vor Abschluss der vollständigen klinischen Prüfung auf den Markt zu bringen. Das Pharmaunternehmen verpflichtet sich in dem Fall, von der Behörde festgelegte Bedingungen innerhalb eines bestimmten Zeitraumes zu erfüllen, beispielsweise vollständige Phase-III-Daten nachzuliefern; die bedingte Zulassung gilt für ein Jahr, ist jedoch jährlich nach Beurteilung durch die Behörde verlängerbar, bis eine reguläre Zulassung erteilt wird. Im Gegensatz zu einer Notfallzulassung ist im Fall einer bedingten Zulassung der Inhaber der Zulassung grundsätzlich in vollem Umfang für das Arzneimittel und seine Sicherheit haftbar.[26][27] Auch dieses Verfahren ist nur nach einer Einzelfallprüfung möglich. Das erste Arzneimittel mit einer bedingten Zulassung in der EU war im Sommer 2006 Sutent (Wirkstoff: Sunitinib). Einer Studie zufolge betrug für in der EU im Zeitraum von 2006 bis 2015 bedingt zugelassene Arzneimittel die Zeit für die Erfüllung der Auflagen im Mittel vier Jahre, bei mehr als einem Drittel der Verfahren gab es in diesem Zusammenhang Verzögerungen oder Unstimmigkeiten. Die Autoren folgern, dass die bedingte Zulassung den Zugang zu Arzneimitteln ermögliche, deren klinischer Wert über längere Zeit nicht vollständig feststehe, mit möglicherweise entsprechenden Risiken für die Patienten.[28]
Das Accelerated approval in den USA hat Gemeinsamkeiten mit der bedingten Zulassung in der EU; auch hier wird eine vorläufige Zulassung erteilt, mit der Auflage, klinische Studien durchzuführen und abzuschließen, in denen ein Patientennutzen mit klinisch relevanten Endpunkten belegt wird. Das Accelerated approval stützt sich bei der Entscheidung im Wesentlichen auf Surrogatmarker. Problematisch ist, dass in den USA Unternehmen den Auflagen, klinische Studien nach der Zulassung abzuschließen, oft nicht nachkommen.[29]
Die Zulassung in Ausnahmefällen (under exceptional circumstances) im zentralisierten EU-Verfahren[30] kommt infrage, wenn für ein Arzneimittel auch in Zukunft wahrscheinlich keine vollständigen Daten über die Wirksamkeit und Sicherheit zur Verfügung gestellt werden können, etwa weil ihre Erfassung nicht möglich oder unethisch ist oder die zu behandelnde Erkrankung sehr selten ist. Die Bedingungen werden jährlich neu beurteilt.
Mit PRIME (priority medicines) bietet die EMA seit 2016 Arzneimittelherstellern ein Vorgehen an, das immer dann gewählt werden kann, wenn es in einem Bereich einen Versorgungsmangel („ungedeckten medizinischen Bedarf“, unmet medical need) gibt, wenn es also an einer wirkungsvollen Therapie fehlt oder das neue Mittel einen therapeutischen Vorteil bietet. Insbesondere kleine Unternehmen oder universitäre Einrichtungen sollen durch kontinuierliche regulatorische und wissenschaftliche Beratung in sehr frühen Entwicklungsphasen gefördert werden. Über 60 Arzneimittel wurden bisher (Stand 2020) in das PRIME-Programm aufgenommen.[31]
Das Konzept der Adaptive Pathways (deutsch etwa „anpassbare Wege“; auch bezeichnet als Medicines Adaptive Pathways to Patients, MAPP) ist ein Teil der Bestrebungen der EMA, innerhalb des bestehenden rechtlichen Rahmens Patienten einen schnelleren Zugang zu neuen Medikamenten durch mehr Flexibilität zu ermöglichen. Das Adaptive-Pathway-Konzept basiert auf drei Prinzipien: Erstens auf einer iterativen Entwicklung – das bedeutet entweder eine schrittweise Zulassung, beginnend mit einer begrenzten Patientenpopulation, die auf eine größere Population ausgeweitet werden kann, oder die Bestätigung eines ausgewogenen Risiko-Nutzen-Verhältnisses eines Produktes nach einer bedingten Zulassung basierend auf frühen Daten mit Surrogatmarkern, die als Prädiktoren für die wichtigen klinischen Outcomes untersucht wurden; zweitens auf dem Sammeln von Evidenz über Versorgungsdaten („real life use“), um damit die Daten aus den klinischen Studien zu ergänzen; drittens auf der frühen Einbindung von Patienten und Bewertungsgremien in die Diskussion über die Entwicklung eines Arzneimittels. Das BfArM und die EMA testeten das Adaptive-Pathway-Vorgehen von 2014 bis 2016 in einem Pilotprojekt.[32][33] Kritiker befürchten Absenkungen von Zulassungsstandards zulasten der bislang streng regulierten Zulassungsaspekte Wirksamkeit und Unbedenklichkeit.[34] Die Adaptive Pathways sollen zunächst nur bei bestimmten Arzneimitteln zur Anwendung kommen, etwa wenn es mangels Behandlungsalternativen einen dringenden Bedarf gibt (unmet medical need).[35] Die Nachverfolgung des Projekts beinhaltet Zusammenarbeit mit Health-Technology-Assessment-Gremien und zusätzlichen Interessenvertretern wie Patienten und Kostenträgern.[32]
Notfallverfahren
Das Rolling-Review-Verfahren („Rolling-Review“ = „fortlaufende Überprüfung“) ist eines der Regulierungsinstrumente, die der Europäischen Arzneimittel-Agentur (EMA) im zentralisierten Zulassungsverfahren zur Verfügung stehen, um die Bewertung eines vielversprechenden Prüfpräparats während eines Notfalls im Bereich der öffentlichen Gesundheit – z. B. einer Pandemie – zu beschleunigen. Die Berichterstatter werden bereits ernannt, während die Entwicklung noch im Gange ist, und die EMA prüft fortlaufend die Daten, sobald sie verfügbar sind.[36]
Für Influenza-Impfstoffe (Grippeimpfstoffe) zur Anwendung in pandemischen Situationen bestehen ebenfalls spezielle Verfahren, um deren Verfügbarkeit zu beschleunigen. Dazu gehören in der EU das Modellimpfstoff-Verfahren (mock-up procedure), mit dem ein bereits zugelassener Modellimpfstoff an den aktuellen pandemischen Virusstamm angepasst wird, sobald dieser identifiziert wurde. Ein weiteres Verfahren ist das „Notfallverfahren“ (emergency procedure). Es ermöglicht die rasche Zulassung eines neuen Influenza-Impfstoffs, wenn ein solcher infolge einer Pandemiemeldung neu entwickelt wird. Für beide Verfahren beträgt die Dauer der Beurteilung durch den wissenschaftlichen Ausschuss 70 statt der sonst üblichen 210 Tage.[37] Ein drittes Verfahren dient der Zulassung von Impfstoffen, die sich von bereits zur Verwendung gegen die saisonale Influenza zugelassenen Impfstoffen ableiten. Diese wurden jedoch derart modifiziert, dass sie zum Schutz gegen pandemische Grippe eingesetzt werden können. Dieses Verfahren wird in der Regel national praktiziert, da die meisten saisonalen Grippeimpfstoffe auf nationaler Ebene zugelassen sind.[37]
In den USA besteht für Arzneimittel die Möglichkeit der Erteilung einer Notfallzulassung (Emergency use authorization, EUA). Eine Notfallzulassung für „medizinische Gegenmaßnahmen“ (Medical countermeasures, MCMs) kann durch die Food and Drug Administration (FDA) ausgesprochen werden, wenn das Vorliegen eines Notfalls im Bereich der öffentlichen Gesundheit festgestellt wurde, beispielsweise im Fall einer CBRN-Bedrohung oder Pandemie. Gesetzliche Grundlage ist der Federal Food, Drug, and Cosmetic Act (FD&C Act).[38]
Orphan-Arzneimittel
Orphan-Arzneimittel sind Arzneimittel, die zur Behandlung, Prävention oder Diagnose einer seltenen Krankheit eingesetzt werden. Seltene Krankheiten in diesem Sinne betreffen in der EU weniger als 5 von 10.000 Personen. In der EU müssen Orphan-Arzneimittel im zentralisierten Verfahren zugelassen werden; der Status Orphan-Arzneimittel wird von der Europäischen Kommission nach einer Empfehlung des Ausschusses für Orphan-Arzneimittel (COMP) der Europäischen Arzneimittelagentur vergeben. Dies ist bis September 2007 für 500 Arzneimittel geschehen; von diesen sind bisher 36 zugelassen worden. Pharmaunternehmen erhalten für Orphan-Arzneimittel von der Europäischen Arzneimittelagentur Unterstützung bei der Planung der erforderlichen Studien, ferner Vergünstigungen bei den Zulassungs- und Inspektionsgebühren und eine zehnjährige Marktexklusivität.[39]
Generika
Generika sind sich im Wesentlichen gleichende Arzneimittel, die denselben Arzneistoff in derselben Dosis und Arzneiform enthalten wie ein nicht mehr patentgeschütztes Referenzarzneimittel. Für diese Arzneimittel gelten vereinfachte Bedingungen zur Zulassung. Dazu muss die Herstellung und pharmazeutische Qualität dokumentiert sowie die Bioverfügbarkeit und Bioäquivalenz zu dem Originalarzneimittel belegt werden. Für die restlichen nichtklinischen und klinischen Daten kann der Antragsteller auf die Daten zum Referenzarzneimittel verweisen. Dies ist in der EU allerdings unabhängig vom Patentschutz erst acht Jahre nach Zulassung des Referenzarzneimittels möglich, die Zulassung selbst wird erst zehn Jahre nach Erteilung der Erstgenehmigung erteilt. Für Biosimilars gelten besondere Bedingungen.
Für Medikamente mit bereits bekannten Wirkstoffen gibt über den „allgemeinen medizinische Gebrauch“ (well-established use) eine weitere Antragsart mit vereinfachten Zulassungsbedingungen: Der Antragsteller muss keine Daten aus eigenen präklinischen und klinischen Prüfungen vorlegen, wenn er nachweisen kann, dass die Wirkstoffe des Arzneimittels seit mindestens zehn Jahren in der EU allgemein medizinisch verwendet wurden. Andere einschlägige wissenschaftliche Dokumentation, aus der die bereits bekannten Wirkungen und Nebenwirkungen ersichtlich sind, ersetzt dann eigene Studien.[40] So können etwa für Arzneimittel mit bekannten pflanzlichen Wirkstoffen beispielsweise die vom Ausschuss für pflanzliche Arzneimittel erarbeiteten Monografien herangezogen werden.
Alternativmedizinische und pflanzliche Arzneimittel
Neben den genannten vereinfachten Registrierungsverfahren für bestimmte Homöopathika und traditionelle pflanzliche Arzneimittel kann in der EU ein Mitgliedstaat für die vorklinischen und klinischen Versuche von zulassungspflichtigen homöopathischen Arzneimittel entsprechend seinen Grundsätzen und den besonderen Merkmalen der homöopathischen Medizin abweichende Vorschriften anwenden.[41][42] In Deutschland ist dies durch die Regelungen des Arzneimittelgesetzes für die Arzneimittel der „besonderen Therapierichtungen“ (neben Homöopathie auch Anthroposophie und Phytotherapie) konkretisiert,[43] wonach es keiner Wirksamkeitsnachweise mittels kontrollierte Studien bedarf.[44] Auch weitere Staaten haben besondere Vorschriften für Homöopathika (siehe Hauptartikel Homöopathisches Arzneimittel #Rechtliche Einordnung)
In der Schweiz regelt eine Verordnung die vereinfachte Zulassung für bestimmte Arzneimittel der Komplementär- und Phytotherapie. Es gelten Sondervorschriften für den Nachweis der therapeutischen Wirksamkeit und Sicherheit.[45]
Verfahren nach erteilter Zulassung
Die Erteilung einer Zulassung ist zwar ein entscheidender Schritt, damit sind aber die regulatorischen Aktivitäten keineswegs beendet. Die Zulassungsunterlagen müssen kontinuierlich aktualisiert werden, und die Anwendung eines Arzneimittels muss ständig überwacht werden. Bei neuen Arzneimitteln wird die Zulassung nur für eine beschränkte Zeitspanne von üblicherweise fünf Jahren erteilt. Vor Ablauf dieser Frist muss die Zulassung erneuert werden, in der Regel gilt sie dann unbegrenzt. Wird jedoch ein genehmigtes Arzneimittel nicht innerhalb einer vorgegebenen Frist in Verkehr gebracht, oder befindet es sich über einen längeren Zeitraum nicht im Markt, so erlischt eine Zulassung in der Regel („Sunset clause“). Für EU-Staaten beträgt diese Frist bzw. Zeitraum drei Jahre.[46][47]
Änderungsanzeigen
Das pharmazeutische Unternehmen hat gegenüber den zuständigen Behörden eine Anzeigepflicht für sämtliche Änderungen, die die erteilte Zulassung betreffen. Je nach Trageweite der Änderungen müssen diese teils von der Behörde genehmigt werden. Einfache, nur anzeigepflichtige Änderungen sind beispielsweise administrative Änderungen beim Hersteller oder kleinere Änderungen im Herstellungsprozess. Zustimmungspflichtig sind beispielsweise Änderungen der Dosis, der Arzneiform oder der Applikationsform. Auch jede Änderung an der Zusammenfassung der Merkmale des Arzneimittels bedarf einer Genehmigung.
Wenn die Zulassung eines Arzneimittels auf eine weitere Indikation ausgeweitet werden soll, erfordert dies einen eigenen, vollständigen Zulassungsantrag.
Pharmakovigilanz
Das pharmazeutische Unternehmen ist verpflichtet, auch nach erteilter Zulassung Erkenntnisse zu unerwünschten Arzneimittelwirkungen zu sammeln und auszuwerten. Der Aufsichtsbehörde ist darüber in vorgegebenen Abständen Bericht zu erstatten, bei schweren, unerwarteten Fällen von Nebenwirkungen auch innerhalb kurzer Fristen. Die fortlaufende Überwachung ist deshalb so wichtig, weil es in klinischen Studien mit nur wenigen Tausend Patienten nicht möglich ist, seltene oder sehr seltene Nebenwirkungen zu erkennen. Auch sehr spät auftretende Nebenwirkungen lassen sich in den Zulassungsstudien nur schwer erfassen.
Neue Erkenntnisse können zur Einschränkung der Zulassung führen, beispielsweise durch eine Änderung der Zusammenfassung der Merkmale des Arzneimittels. Ergeben sich während der Anwendung eines Arzneimittels Erkenntnisse zu schwerwiegenden Nebenwirkungen, die das Nutzen-Risiko-Verhältnis ungünstig werden lassen, kann eine Zulassung auch vollständig widerrufen werden. Von den zwischen 1975 und 1999 in den USA zugelassenen Arzneimitteln mit neuen Wirkstoffen wurden 2,9 % wegen Mängeln vom Markt genommen.[48] Im Vereinigten Königreich waren es im Zeitraum von 1972 bis 1994 3,8 %.[49] Der weit überwiegende Grund für die Marktrücknahme sind dabei nicht vertretbare Nebenwirkungen. In manchen Fällen, wie beispielsweise bei Clobutinol, erfolgte eine Widerrufung der Zulassung aufgrund von Sicherheitsbedenken erst nach jahrzehntelanger Verwendung des Arzneimittels.
Auch im Bereich der Pharmakovigilanz prüfen die Arzneimittelbehörden durch Inspektionen bei den Pharmaunternehmen, ob die vorgeschriebenen Überwachungsmaßnahmen korrekt umgesetzt werden.
Geschichte der Arzneimittelzulassung
Zulassungsverfahren wurden weltweit eingeführt, nachdem die Arzneimittelzubereitung in Apotheken durch industrielle Fertigung weitgehend verdrängt wurde und verschiedene Arzneimittelskandale das Vertrauen in die Arzneimittelsicherheit erschüttert hatten.
Seit Beginn des 20. Jahrhunderts hatte die industrielle Fertigung von Arzneimitteln die Apothekenzubereitung weitgehend verdrängt. Dennoch hatten nur wenige Länder schon in der ersten Hälfte des zwanzigsten Jahrhunderts Marktzulassungsverfahren für Arzneimittel, darunter Frankreich, Schweden, Norwegen und vor allem die USA.[50] In den USA wurde eine Zulassung von neuen Arzneimitteln bereits durch den Federal Food, Drug and Cosmetic Act von 1938 zur Pflicht; dieses Gesetz wurde nach der Sulfanilamid-Katastrophe vom Kongress verabschiedet. Allerdings beschränkten sich die Zulassungskriterien damals auf die pharmazeutische Qualität und Unbedenklichkeit; ein Arzneimittel galt damals als zugelassen, wenn die zuständige Behörde, die Food and Drug Administration (FDA) nicht innerhalb einer bestimmten Frist widersprach.
Das Wirksamkeitskriterium und die heutige Zulassungsprozedur wurden in den USA erst 1962 durch das Kefauver-Harris Drug Amendment eingeführt, das zeitgleich mit der Aufdeckung des Contergan-Skandals beraten wurde. Die damaligen Ereignisse haben den Gesetzgebungsprozess nachhaltig beeinflusst; aus dem ursprünglichen Gesetzgebungsverfahren im Kongress gegen zu hohe Arzneimittelpreise und unlautere Arzneimittelwerbung wurde so ein Gesetz zum Verbraucherschutz. Die damals in den USA entwickelten restriktiven Zulassungskriterien wurden in den folgenden Jahren von vielen anderen Ländern übernommen. Ende der 1980er und Anfang der 1990er Jahre wurden in den USA anlässlich des dringenden Bedarfs an Arzneimitteln zur Behandlung von Aids beschleunigte Zulassungsverfahren eingeführt.
Ebenfalls als Konsequenz des Contergan-Skandals wurde in der Europäischen Wirtschaftsgemeinschaft die Richtlinie 65/65/EWG des Rates vom 26. Januar 1965 zur Angleichung der Rechts- und Verwaltungsvorschriften über Arzneispezialitäten verabschiedet. Diese sah erstmals eine Genehmigung für das Inverkehrbringen von Arzneimitteln vor und forderte einen Nachweis der therapeutischen Wirksamkeit. Die folgende Richtlinie 75/319/EWG von 1975 war deutlich detaillierter in den Zulassungsanforderungen. Außerdem wurde mit der Richtlinie ein neues europäisches Expertengremium gegründet, der Ausschuss für Arzneispezialitäten (engl. Committee for Proprietory Medicinal Products, CPMP), einem Vorläufer des heutigen Ausschusses für Humanarzneimittel (CHMP). 1987 wurde mit der Richtlinie 87/22/EWG das Konzertierungsverfahren für innovative Arzneimittel definiert, einem Vorläufer des heutigen zentralisierten Verfahrens, das 1993 mit der Richtlinie 2309/93/EWG definiert und ab 1995 umgesetzt wurde. Erst mit den 1995 in Kraft getretenen Reformen fanden die Gemeinschaftsverfahren breite Anwendung. Im Review der europäischen Arzneimittelgesetzgebung von 2001 bis 2004 wurde dann der heutige Rahmen geschaffen.[51]
In der Bundesrepublik Deutschland war die Umsetzung der ersten EWG-Richtlinie von 1965 in nationales Recht ein langwieriger Prozess, der erst mit dem Inkrafttreten des zweiten Arzneimittelgesetzes von 1976 seinen Abschluss fand. Erst 1975 wurde im damaligen Bundesgesundheitsamt ein Institut für Arzneimittel für die Arzneimittelzulassung gegründet, nach Auflösung des Bundesgesundheitsamtes 1994 in der Folge des Blut-Aids-Skandales wurde daraus das heutige Bundesinstitut für Arzneimittel und Medizinprodukte. Die Anwendung der neuen Rechtsinstrumente auf alte Arzneimittel im Nachzulassungsverfahren dauerte in Deutschland noch bis Ende 2005.[52]
Literatur
- Martin Lorenz: Das gemeinschaftliche Arzneimittelzulassungsrecht unter besonderer Berücksichtigung der Reform 2004/2005. Nomos-Verlags-Gesellschaft, Baden-Baden 2006, ISBN 3-8329-1912-0.
Weblinks
- EudraLex - Volume 2 - Pharmaceutical legislation on notice to applicants and regulatory guidelines for medicinal products for human use
- Europäische Arzneimittelagentur (European Medicines Agency)
- Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM)
- Datenbank der zugelassenen Arzneimittel beim BfArM
- Paul-Ehrlich-Institut
- Österreichische Agentur für Gesundheit und Ernährungssicherheit
- Swissmedic (Schweizerisches Heilmittelinstitut)
- Arzneimittel in der Europäischen Union (PDF; 1,2 MB)
Einzelnachweise
- Elizabeth Storz: Arzneimittelsicherheit. In: GIT Labor-Fachzeitschrift. August 2008, S. 712–714.
- § 4 Abs. 15 AMG.
- Chemistry Manufacturing and Controls (CMC). Guidances for Industry (GFIs). Abgerufen am 16. März 2021.
- Karl Feiden, Hermann Pabel: Wörterbuch der Pharmazie. Band 3: Arzneimittel- und Apothekenrecht. Wissenschaftliche Verlagsgesellschaft, Stuttgart 1985, ISBN 3-8047-0670-3.
- Richtlinie 2001/83/EG, Anhang I Teil I Absatz 5.2.5.1.
- Robert Kemp und Vinay Prasad: Surrogate endpoints in oncology: when are they acceptable for regulatory and clinical decisions, and are they currently overused?, BMC Medicine Band 15, Artikel 134 (2017), abgerufen 30. Oktober 2019.
- STIKO: Methoden zur Durchführung und Berücksichtigung von Modellierungen zur Vorhersage epidemiologischer und gesundheitsökonomischer Effekte von Impfungen für die Ständige Impfkommission, 16. März 2016, abgerufen 30. Oktober 2019.
- FDA: Table of Surrogate Endpoints That Were the Basis of Drug Approval or Licensure, online 30. September 2019, abgerufen 30. Oktober 2019.
- BVerwGE Band 84, S. 215–224, Urteil vom 14. Oktober 1993 Az. 3 C 21.91.
- Kuss O, Blettner M, Börgermann J: Propensity Score – eine alternative Methode zur Analyse von Therapieeffekten. In: part 23 of a series on evaluation of scientific publications. Dtsch Arztebl Int 2016; 113: 597–603. DOI: 10.3238/arztebl.2016.0597, 5. September 2016, abgerufen am 17. Dezember 2021.
- § 5 AMG.
- Dieter Hart: Die Nutzen/Risiko-Abwägung im Arzneimittelrecht. Ein Element des Health Technology Assessment. In: Bundesgesundheitsblatt. Band 48, 204-14 (2005), PMID 15726462. doi:10.1007/s00103-004-0977-2
- P. Dieppe, C. Bartlett, P. Davey, L. Doyal, S. Ebrahim: Balancing benefits and harms: the example of non-steroidal anti-inflammatory drugs. In: BMJ. Band 329(7456), 2004, S. 31–34, PMID 15231619.
- Wissenschaftlicher Beirat des Paul-Ehrlich-Instituts: Protokoll der konstituierenden Sitzung am 30. Mai 2017 (PDF).
- Zentralisiertes Verfahren auf der Website des Bundesinstituts für Arzneimittel und Medizinprodukte, abgerufen am 4. November 2021.
- Volume 2A - Procedures for marketing authorisation, Chapter 2 - Mutual Recognition. (PDF) In: EudraLex - Volume 2 - Pharmaceutical legislation on notice to applicants and regulatory guidelines for medicinal products for human use (englisch).
- Statistiken zu nicht zentralisierten Zulassungsverfahren bei den Heads of Medicines Agencies.
- Information für Pharmaunternehmen, auf der Website der FDA (englisch).
- Opinion | The Solution to Drug Prices. 9. September 2015 (nytimes.com [abgerufen am 29. Oktober 2018]).
- International Cooperation in Pharmaceuticals - Key documents, EU-Kommission. Abgerufen am 10. Juni 2020.
- Accelerated assessment im Glossar der EMA, abgerufen am 10. Juni 2020.
- NME Drug and New Biologic Approvals in 2006, FDA Dokumentenarchiv, abgerufen am 10. Juni 2020.
- New Molecular Entity (NME) Drug and New Biologic Approvals, FDA Website, abgerufen am 10. Juni 2020.
- EMEA concludes first accelerated assessment for a medicine for human use, EMA-Pressemitteilung vom 27. April 2007 (PDF).
- Conditional marketing authorisation im Glossar der EMA, abgerufen am 10. Juni 2020.
- Questions and Answers: Conditional Marketing Authorisation of COVID-19 Vaccines in the EU. Europäische Kommission, 11. Dezember 2020, abgerufen am 20. August 2021 (englisch).
- Fragen und Antworten: Bedingte Marktzulassung für COVID-19-Impfstoffe in der EU. Europäische Kommission, 11. Dezember 2020, abgerufen am 20. August 2021.
- R. Banzi, C. Gerardi, V. Bertele, S. Garattini: Approvals of drugs with uncertain benefit–risk profiles in Europe. In: European Journal of Internal Medicine. Band 26, 2015, S. 572–584, doi:10.1016/j.ejim.2015.08.008.
- C. D. Furberg, A. A. Levin, P. A. Gross, R. S. Shapiro, B. L. Strom: The FDA and drug safety: a proposal for sweeping changes. In: Arch Intern Med. Band 166(18), 2006, S. 1938–1942, PMID 17030825.
- Exceptional circumstances im Glossar der EMA, abgerufen am 10. Juni 2020.
- European Medicines Agency: PRIME: priority medicines. Abgerufen am 12. Juni 2020.
- EMA: Adaptive pathways: ‘The adaptive pathways approach is part of the European Medicines Agency’s (EMA) efforts to improve timely access for patients to new medicines’, abgerufen am 10. September 2017.
- Final report on the adapt ive pathways pilot Summary des Pilotprojektes vom 28. Juli 2016, abgerufen am 10. September 2017.
- P.V. Bonanno et al.: Adaptive Pathways: Possible Next Steps for Payers in Preparation for Their Potential Implementation. In: Frontiers in Pharmacology. Band 8, 2017, doi:10.3389/fphar.2017.00497, PMC 5572364 (freier Volltext).
- H.G. Eichler et al.: Medicines Adaptive Pathways to Patients: Why, When, and How to Engage? In: Clinical Pharmacology and Therapeutics. Band 105, Nr. 5, 2018, doi:10.1002/cpt.1121, PMC 6585618 (freier Volltext).
- European Medicines Agency: How EMA fast-tracks development support and approval of medicines and vaccines. Abgerufen am 12. Juni 2020.
- European Medicines Agency (EMA): Authorisation Procedures. Abgerufen am 12. Juni 2020.
- FDA: Emergency Use Authorization. Abgerufen am 27. Juni 2020.
- Europäische Arzneimittelagentur zu Orphan-Arzneimitteln (Medicines for rare diseases). (Memento vom 31. Januar 2017 im Internet Archive) (englisch).
- Well-established use im Glossar der EMA, abgerufen am 10. Juni 2020.
- National implementation European Coalition on Homeopathic & Anthroposophic Medicinal Products, 3. Dezember 2020. Abgerufen am 17. Dezember 2021.
- Richtlinie 2001/83/EG des europäischen Parlament und des Rates vom 6. November 2001, Artikel 16 (2).
- Besondere Therapierichtungen und Traditionelle Arzneimittel, BfArM, abgerufen am 17. Dezember 2021.
- 4. Besondere Therapierichtungen und traditionelle Arzneimittel. Stiftung für Qualität und Wirtschaftlichkeit im Gesundheitswesen, rechtsfähige Stiftung des bürgerlichen Rechts, abgerufen am 15. Dezember 2021 (deutsch).
- Verordnung des Schweizerischen Heilmittelinstituts über die vereinfachte Zulassung und das Meldeverfahren von Komplementär- und Phytoarzneimitteln, Institutsrat des Schweizerischen Heilmittelinstituts. Abgerufen am 17. Dezember 2021.
- Richtlinie 2001/83/EG des Europäischen Parlaments und des Rates vom 6. November 2001 zur Schaffung eines Gemeinschaftskodexes für Humanarzneimittel, Artikel 24.
- Verordnung (EG) Nr. 726/2004, Artikel 14.
- K. E. Lasser, P. D. Allen, S. J. Woolhandler, D. U. Himmelstein, S. M. Wolfe, D. H. Bor: Timing of new black box warnings and withdrawals for prescription medications. In: JAMA. Band 287(17), 2002, S. 2215–2220, PMID 11980521.
- D. B. Jefferys, D. Leakey, J. A. Lewis, S. Payne, M. D. Rawlins: New active substances authorized in the United Kingdom between 1972 and 1994. In: Br J Clin Pharmacol. Band 45(2), 1998, S. 151–156, PMID 9491828.
- Jürgen Feick: Marktzugangsregulierung: Nationale Regulierung, europäische Integration und internationale Harmonisierung in der Arzneimittelzulassung. In: Roland Czada, Susanne Lütz (Hrsg.): Die politische Konstitution von Märkten. Westdeutscher Verlag, Wiesbaden 2000, ISBN 3-531-13415-9, S. 228–249.
- Jürgen Feick: Marketing authorization for pharmaceuticals in the European Union. In: Joining-up Europe’s Regulators. European Policy Forum, London 2008, ISBN 978-1-903850-29-9, S. 35–63.
- Reinhard Kurth: Die Entwicklung des Bundesinstituts für Arzneimittel und Medizinprodukte (BfArM) im zunehmenden europäischen Wettbewerb. In: Bundesgesundheitsblatt. Band 51(3), 2008, S. 340–344, PMID 18369569.