Enantiomer
Enantiomere sind Stereoisomere chemischer Verbindungen, die sich in ihrer Konstitution decken und sich in den räumlichen Strukturen zu einem Gegenstück verhalten wie dessen (nicht-deckungsgleiches) Spiegelbild. Man nennt sie aufgrund dieser Tatsache auch Spiegelbildisomere. Die griechische Namensgebung lässt diese Bedeutung erkennen: ἐνάντιος, Gegenstück, und μέρος, Teil oder Bereich. Die Summenformel und die Verknüpfung der jeweiligen atomaren Formationen stimmen überein. Es handelt sich um eine Form der Konfigurationsisomerie; Enantiomere können, im Gegensatz zu Konformationsisomeren, nicht durch Drehung von Atombindungen zur Deckung gebracht werden. Da Enantiomere in allen Stereozentren jeweils die entgegengesetzte Konfiguration besitzen, gibt es theoretisch immer ein (−)- und ein (+)-Enantiomer, von denen in der Natur aber oftmals nur eines vorhanden ist.
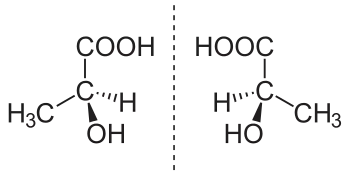
Links: (S)-Milchsäure, rechts: (R)-Milchsäure
Physikalische und chemische Eigenschaften
_(S)-Lactic_Acid.svg.png.webp)
Diese Art der Isomerie wird als Chiralität (Händigkeit) bezeichnet. Zur Veranschaulichung der Spiegelbildlichkeit von Enantiomeren lassen sich Körperteile wie linke und rechte Hand oder Alltagsgegenstände wie linker und rechter Schuh sowie links- und rechtsdrehende Schrauben oder Muttern heranziehen. Ein Beispiel für ein Molekül, das in zwei räumlichen Varianten vorkommt, ist die Milchsäure im Joghurt. Enantiomere haben immer in sämtlichen Stereozentren die entgegengesetzte Konfiguration. Dem gegenüber stehen die Diastereomere, bei denen immer mindestens ein Stereozentrum von mehreren gleich und mindestens eines verschieden konfiguriert ist (siehe dort).
Enantiomere besitzen mit Ausnahme der optischen Aktivität gleiche physikalische Eigenschaften wie Schmelz- und Siedepunkte, Dichte, Löslichkeit, IR-Spektren, Röntgenbeugungsspektren usw. Sie sind optisch aktiv, drehen also die Polarisationsebene des linear polarisierten Lichts im Uhrzeigersinn (rechtsdrehende Form, auch (+)-Form oder früher d-Form genannt) oder gegen den Uhrzeigersinn (linksdrehende Form, (−)-Form oder früher l-Form genannt). Der Drehsinn ist dabei bezüglich der Blickrichtung des Beobachters zu verstehen, nicht bezüglich der Strahlrichtung. Enantiomere drehen die Polarisationsebene des linear polarisierten Lichts um den gleichen Betrag in entgegengesetzte Richtung.
Die beiden Enantiomere eines Eduktes reagieren in chemischen Reaktionen, bei denen ein anderer enantiomerenreiner Reaktionspartner beteiligt ist, unterschiedlich. Die Reaktionsübergangszustände sind dann diastereomer zueinander. Auch beim Einsatz als Arzneistoff in Organismen rufen zueinander enantiomere Stoffe unterschiedliche Wirkungen hervor. Dies lässt sich mit einem Beispiel aus dem Alltag veranschaulichen, dem Anziehen von Schuhen: Es ist klar, dass nur der rechte Schuh zum rechten Fuß passt. Versucht man den rechten Schuh auf den linken Fuß zu ziehen, so wird man damit scheitern oder nur ein sehr dürftiges Ergebnis erzielen. Man erreicht also damit anstelle einer erwünschten Wirkung ein nutzloses oder schädliches und somit unerwünschtes Ergebnis.
Bedeutung für die biologische Wirkung
Viele biologisch wichtige Substanzen sind chiral, nicht nur die kleineren Moleküle von Aminosäuren und Zuckern, sondern auch biologische Makromoleküle wie Enzyme oder Rezeptoren. Bei einigen Substanzklassen überwiegt oft ein Enantiomer, so liegen beispielsweise fast alle natürlichen Aminosäuren in der L-Form vor. Die D-Form ist bei den natürlichen Zuckern (z. B. D-Glucose) überaus dominant, L-Zucker sind rare Exoten. Chiralität als Folge des räumlichen Baus von Molekülen hat entscheidende Bedeutung für das Funktionieren biologischer Systeme, die alle selbst chiral sind. So sind viele Enzymreaktionen auf ein Enantiomer, entweder das linksdrehende oder das rechtsdrehende, spezialisiert: Die Reaktionsgeschwindigkeit mit dem spiegelbildlichen Enantiomer als Substrat ist deutlich geringer oder es wird gar nicht umgesetzt, da das aktive Zentrum eines Enzyms vielfach das eine Enantiomer leichter aufnehmen kann als das andere (Schlüssel-Schloss-Prinzip, Substratspezifität).
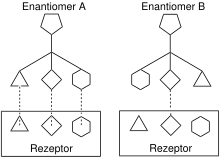
Die Substratspezifität lässt sich durch das Dreipunkt-Interaktionskonzept für Enantiomere veranschaulichen, wie es im bildlichen Schema dargestellt ist: das chirale Enantiomer A ist kongruent zum Rezeptor. Allerdings ist das Spiegelbild von Enantiomer A, das Enantiomer B nicht passend, was zu Bindungsproblemen führen kann und sich somit auf die Wirkung eines Stoffes (beispielsweise eines Arzneistoffes) auswirkt. Es können Unterschiede in der Pharmakodynamik oder der Pharmakokinetik auftreten. Das Enantiomer mit der höheren Aktivität bzw. Affinität wird Eutomer und das mit der niedrigeren Aktivität bzw. Affinität Distomer genannt.[1]


Gar nicht so selten entfaltet das „falsche“ Enantiomer auch eine völlig andere biologische Wirkung. Beispiele:
- Geschmack: Die Aminosäure (S)-Valin schmeckt bitter, (R)-Valin schmeckt süß,
- Geruch: Das Terpen (S)-(+)-Carvon riecht nach Kümmel, dessen Enantiomer (R)-(−)-Carvon riecht nach Minze.[2]
- Pharmakologische Wirkung von Betablockern: Bei Betablockern wirkt das (S)-Enantiomer selektiv auf das Herz, das (R)-Enantiomer hingegen an den Zellmembranen des Auges. Deshalb ist eine hohe Enantiomerenreinheit bei vielen Arzneistoffen von großer Bedeutung.[3]
- Pharmakologische Wirkung von Thalidomid: Die Öffentlichkeit ist durch den Contergan-Skandal auf die fruchtschädigende Wirkung des Thalidomids aufmerksam geworden. Früh wurde dies mit der unterschiedlichen Wirkung der beiden Enantiomere ein und derselben Substanz in Verbindung gebracht, da allein das (S)-Enantiomer des Thalidomids eine teratogene (fruchtschädigende) Wirkung habe, das (R)-Enantiomer jedoch nicht.
Die Thalidomid-Enantiomere weisen jedoch die Eigenschaft auf, dass sie sich im Körper innerhalb von ca. acht Stunden[4] ineinander umwandeln (racemisieren).[5] Die Einnahme ausschließlich (R)-konfigurierten Thalidomids bleibt somit in der Praxis bedeutungslos.[6]
Bei der synthetischen Herstellung von unterschiedlich wirkenden enantiomeren Wirkstoffen etwa in der Pharmakologie versucht man heute, von vornherein nur noch das Enantiomer mit der gewünschten Wirkung herzustellen und als pharmazeutischen Wirkstoff einzusetzen, während man das andere Enantiomer mit seiner möglicherweise unerwünschten – bis hin zur toxischen – Wirkung von Anfang an ausschließen möchte (enantioselektive Synthese). Alternativ kann ein Racemat (1:1-Mischung von zwei Enantiomeren) der Racematspaltung unterworfen werden, um einen einheitlichen (= enantiomerenreinen) Pharmawirkstoff zu gewinnen, der dann mit viel höherer Selektivität als das Racemat pharmakologisch wirksam werden kann.
Auch Geruch oder Geschmack von Stoffen können je nach Enantiomer unterschiedlich ausfallen, weil die entsprechenden Rezeptoren im Körper stets selbst chiral (genauer: enantiomerenrein) sind.
Enantiomere werden in biologischen Systeme in der Regel unterschiedlich metabolisiert.
Racemat
Ein 1:1-Gemisch von (+)- und (−)-Form eines Stoffes, bei dem sich die optische Aktivität der einzelnen Stoffe ausgleicht, nennt man ein Racemat, z. B. (±)-Methionin [Synonyme: DL-Methionin und (RS)-Methionin]. Es ist optisch nicht aktiv und hat einen Drehwinkel α von 0°, da sich die Anteile rechtsdrehender und linksdrehender Form gerade aufheben. Aus dem Quotienten des gemessenen Drehwinkels zum maximalen Drehwinkel des reinen Enantiomers multipliziert mit 100 ergibt sich die optische Reinheit (%) des Enantiomerengemisches. Unter der Annahme idealen Verhaltens (keine Wechselwirkung zwischen den beiden Enantiomeren und Gültigkeit des Lambert-Beerschen Gesetzes) ist die optische Reinheit gleich dem Enantiomerenüberschuss ee.
Der Schmelzpunkt eines Racemats weicht in der Regel vom Schmelzpunkt der reinen Enantiomere ab.[7] Dabei kann der Schmelzpunkt des Racemats tiefer oder höher liegen als der der reinen Enantiomeren. Dieses auf den ersten Blick unerwartete Phänomen kann erklärt werden: Wenn das Racemat als racemisches Gemisch (Konglomerat) kristallisiert, liegen die Kristalle der (+)- und (−)-Form getrennt nebeneinander vor, d. h., das (+)-Enantiomer hat eine höhere Affinität zu (+)-Molekülen und das (−)-Enantiomer hat eine höhere Affinität zu (−)-Molekülen. Es entstehen also beim Kristallisieren „nebeneinander“ reine (+)- und (−)-Kristalle. Der Schmelzpunkt des „racemischen Gemischs“ liegt deutlich unter dem Schmelzpunkt der reinen Enantiomere. Beispiel: Beide reinen (+)- und (−)-Enantiomere des Arzneistoffes Glutethimid schmelzen bei 102–103 °C. Hingegen hat (±)-Glutethimid, also das racemische Gemisch, einen Schmelzpunkt von 84 °C.
Anders ist die Situation, wenn die (+)-Enantiomere beim Kristallisieren bevorzugt mit den (−)-Enantiomeren zusammenlagern. Dann enthält „jeder“ Kristall gleich viele Moleküle „beider“ Enantiomere. Man nennt diesen Fall eine racemische Verbindung. Die racemische Verbindung unterscheidet sich in ihren physikalischen Eigenschaften von den reinen Enantiomeren. Der Schmelzpunkt kann höher, gleich oder niedriger liegen als der der reinen Enantiomeren. Beispiel: Die reinen Enantiomeren des Arzneistoffes Ibuprofen haben einen Schmelzpunkt bei 50–52 °C, racemisches Ibuprofen hat einen Schmelzpunkt bei 75–77,5 °C. Racemisches Ibuprofen kristallisiert also als racemische Verbindung.

R- und S-Sequenzregel (CIP-Regel)
- Enantiomere werden nach der R- und S-Sequenzregel eingestuft.
- Um herauszufinden, ob ein Enantiomer die (R)- oder (S)-Konfiguration besitzt, muss man alle Substituenten nach ihrer Priorität ordnen: 1>2>3>4. Den Substituenten mit der niedrigsten Priorität (4) dreht man unter die Papierebene. Nun geht man von 1 über 2 nach 3.
- Wenn die Richtung, in der man sich bewegt, mit dem Uhrzeigersinn verläuft, ist das Enantiomer (R)-konfiguriert (aus lat. rectus ‚rechtmäßig, richtig, rechts‘)[8]
- Wenn die Richtung, in der man sich bewegt, gegen den Uhrzeigersinn verläuft, ist das Enantiomer (S)-konfiguriert (aus lat. sinister ‚links‘)
- Siehe auch: Cahn-Ingold-Prelog-Konvention für eine Erklärung, wie man die Substituenten nach Prioritäten ordnet.
Aus dem Uhrzeigersinn der sich beim Abzählen der Prioritäten der Substituenten zur Festlegung der Konfiguration [(R) oder (S)] ergibt kann nicht automatisch auf den Drehwinkel α oder die Drehrichtung [(+) oder (−)] der Polarisationsebene des linear polarisierten Lichts geschlossen werden. Beispiele:
Nomenklatur
Zu Unterscheidung der Enantiomere bedient man sich der CIP-Konvention (Cahn-Ingold-Prelog-Konvention, auch R-S-Nomenklatur), mit der die räumliche Anordnung der Substituenten beschrieben wird. Bei bestimmten Substanzklassen (Zucker, begrenzt auf die Biochemie auch bei Aminosäuren) wird nach wie vor die ältere Fischer-Projektion (D,L-Nomenklatur) benutzt, die den Vorteil hat, dass die Bezeichnungen von verwandten Verbindungen gleich sind. Im Namen einer Verbindung kann man die Drehrichtung des Lichtes durch Voransetzen von „(+)-“ für rechtsdrehend beziehungsweise „(−)-“ für linksdrehend deutlich machen; z. B. (−)-Weinsäure oder (+)-Milchsäure, diese Beschreibung ist jedoch nicht immer eindeutig, weil das verwendete Lösungsmittel den Drehsinn in einigen Fällen beeinflussen und damit auch ändern kann.
Oft wird die Vorsilbe Levo- oder Lev- (links) für linksdrehende und Dex- bzw. Dextro- (rechts) für rechtsdrehende Substanzen verwendet.
Beispiele:
- Levodopa, Levothyroxin, Levonorgestrel, Levofloxacin, Levobupivacain, Levetiracetam, Levocetirizin
- Dextrose, Dexamfetamin, Dexibuprofen, Dexketoprofen, Dextromethorphan, Dexrazoxan, Dexchlorpheniramin
Von der CIP-Nomenklatur abgeleitet können (S)-Enantiomere von Arzneistoffen die Vorsilbe Es- und (R)-Enantiomere die Vorsilbe Ar- tragen, wenn es bereits einen Freinamen für die racemische Substanz gibt. Umgekehrt wird gelegentlich für die Bezeichnung von Racematen dem Trivial- oder Enantiomernamen das Präfix Rac- vorangestellt.
Beispiele:
- Armodafinil, Arhalofenat
- Eszopiclon, Esomeprazol, Escitalopram, Esketamin
- Racecadotril, Racepinephrin
Geschichte
Im Jahre 1848 gelang Louis Pasteur die Racematspaltung für die Enantiomere eines Salzes der D- und L-Weinsäure. Sie unterschieden sich für ihn lediglich darin, dass ihre Kristalle spiegelbildlich aufgebaut waren. Nach sorgfältiger Kristallisation konnte er die verschiedenen Kristalle in mühevoller Handarbeit trennen und leitete damit die Erforschung der Enantiomerie ein. Auch bei der Zusammenführung von optischer Aktivität einer Substanz und der absoluten Konfiguration der Moleküle durch Johannes Martin Bijvoet spielte die Weinsäure eine wichtige Rolle. Natrium-Rubidium-Tartrat (ein Salz der Weinsäure) spielte eine zentrale Rolle bei der zuverlässigen Aufklärung der absoluten Konfiguration von enantiomerenreinen Molekülen. Die Entdecker wurden dafür mit dem Nobelpreis für Chemie ausgezeichnet.
Chemie
Die synthetische Chemie verfügt neuerdings über Methoden zur direkten gezielten Herstellung eines reinen Wirkstoffisomers durch enantioselektive oder gar enantiospezifische Synthesen nach dem Vorbild der Natur.
Asymmetrische Synthese
Bei chemischen Synthesen chiraler Stoffe entstehen meist beide Enantiomere im gleichen Verhältnis. Sie müssen aufwendig getrennt werden, um die Enantiomere als Reinstoff zu erhalten. Die Synthese enantiomerenreiner Moleküle gehört zu den schwierigsten Feldern der präparativen Organischen Chemie. Einen Ausweg bieten hier zahlreiche neuere Syntheseverfahren, die zum Teil sehr große Enantioselektivitäten aufweisen. Um ein chirales Molekül aus nicht-chiralen Edukten zugänglich zu machen, wurden verschiedene Methoden entwickelt:
- Verwendung chiraler Hilfsreagenzien und Katalysatoren (z. B. chirale Phosphane)
- Umsetzung mit Enzymen
- Anbringung eines Auxiliars, das nach der Reaktion wieder entfernt werden kann
- Überführen in Diastereomere [durch Anbringung eines enantiomerenreinen Substituenten wie (−)-Strychnin] und deren Trennung (z. B. Kristallisation, Säulenchromatographie u. a.)
Die hierbei erreichte Enantiomerenreinheit ist oft unterschiedlich hoch. Als Maß für den Erfolg der asymmetrischen Synthese/Kristallisation wird der Enantiomerenüberschuss angegeben:

Daneben bleibt die Synthese von enantiomerenreinen Wirkstoffen aus chiralen Naturstoffen (Beispiele: Aminosäuren, Kohlenhydrate, Terpene, Alkaloide, Steroide) eine wichtige und effiziente Methode.
Arzneistoffsynthese
Als wichtiger Wegbereiter für die gezielte enantiomerenreine Arzneistoffsynthese gilt der Pharmakologe Everhardus Ariëns, der bereits in den 1980er Jahren racemische Arzneistoffe als enantiomer verunreinigte Wirksubstanzen beanstandete. Mit der Weiterentwicklung der synthetischen Chemie ist die stereoselektive vollsynthetische Produktion reiner Enantiomere mit einem bis wenigen Chiralitätszentren heutzutage häufig ohne größeren Aufwand möglich. Arzneistoffe mit vielen Asymmetriezentren hingegen werden ausgehend von Naturstoffen bis auf wenige Ausnahmen teilsynthetisch hergestellt.[10]
Unter den monochiralen Arzneistoffen ist ein Trend zur Synthese von enantiomerenreinen Substanzen erkennbar. Waren 1999 bis 2003 unter insgesamt 24 Neueinführungen bereits 15 reine Enantiomere zu finden, stieg die Zahl für den Zeitraum 2004 bis 2008 auf 20 reine Enantiomere unter 25 Neueinführungen. Größtenteils handelt es sich um völlig neue Stoffe, also Enantiomere, die keine Racemat-Vorläufer haben.[10] In den folgende beiden 5-Jahreszeiträumen war der Anteil enantiomerenreiner Neuentwicklungen weiterhin hoch.[11][12]
Einige enantiomere Arzneistoffe wurden indessen entwickelt, um als Eutomer umsatzstarke Racemate zu ersetzen. Nicht immer jedoch bietet ein enantiomerenreiner Stoff einen echten therapeutischen Vorteil gegenüber dem Racemat.[13] So ist es zweifelhaft, ob das 2001 eingeführte pharmakologisch aktive Dexibuprofen [(S)-Enantiomer des racemischen Ibuprofens] ein wirklicher Fortschritt ist, da das (R)-Enantiomer des Ibuprofens nach Resorption sowieso rasch enzymatisch in die wirksame (S)-Form umgewandelt wird. Sowohl der enantiomerenreine, als auch der racemische Arzneistoff sind laut Herstellerempfehlung gleich zu dosieren. Auch für den 2000 eingeführten Hustenblocker Levodropropizin ist die therapeutische Überlegenheit gegenüber der Verwendung des Racemats fragwürdig, da keine eindeutige Unterscheidung in ein wirksames und unwirksames Enantiomer bekannt wurde und beide ähnlich dosiert werden. Der ebenfalls 2000 eingeführte Protonenpumpenhemmer Esomeprazol wirkt als Prodrug der achiralen Wirkform zwar nicht stärker als die (R)-Form des Omeprazols, ist aber aufgrund einer langsameren enzymatischen Verstoffwechselung besser bioverfügbar. Dennoch ist aufgrund des Wirkungsmechanismus der Protonenpumpenhemmer die therapeutische Relevanz in Frage gestellt worden.[13]
Als therapeutischer Fortschritt im Vergleich zur Wirksamkeit der Vorgänger-Racemate hingegen werden beispielsweise die Substanzen Escitalopram, Levocetirizin, Levobupivacain und Dexrazoxan angesehen.
Bei manchen Stoffen liegt das Chiralitätszentrum nicht im pharmakologisch aktiven Molekülbereich, wie etwa bei den Gyrasehemmern Gatifloxacin oder Nadifloxacin. Dann sollte die Verwendung des Racemats vertretbar sein.[13]
Literatur
- E. J. Ariëns: Stereochemistry, a basis for sophisticated nonsense in pharmacokinetics and clinical pharmacology. In: European Journal of Clinical Pharmacology. Band 26, Nr. 6, 1984, S. 663–668, doi:10.1007/BF00541922.
- Adam Sobanski, Roland Schmieder, Fritz Vögtle: Topologische Stereochemie und Chiralität. In: Chemie in unserer Zeit. Band 34, Nr. 3, 2000, S. 160–169, doi:10.1002/1521-3781(200006)34:3<160::AID-CIUZ160>3.0.CO;2-6.
- Klaus Roth: Eine unendliche chemische Geschichte. In: Chemie in unserer Zeit. Band 39, Nr. 3, 2005, S. 212–217, doi:10.1002/ciuz.200590038.
- Bernard Testa: Grundlagen der Organischen Stereochemie. Wiley-VCH, 1983, ISBN 3-527-25935-X.
- Uwe Meierhenrich: Amino acids and the asymmetry of life. Springer-Verlag, 2008, ISBN 978-3-540-76885-2.
Einzelnachweise
- H. J. Roth, C. E. Müller, G. Folkers (Hrsg.): Stereochemie & Arzneistoffe: Grundlagen – Betrachtung – Auswirkung, wissenschaftliche Verlagsgesellschaft mbH, Stuttgart, S. 80, 81, 1998.
- Wolfgang Legrum: Riechstoffe, zwischen Gestank und Duft, Vieweg + Teubner Verlag (2011) S. 33–35, ISBN 978-3-8348-1245-2.
- Everhardus Ariëns: Stereochemistry, a basis for sophisticated nonsense in pharmacokinetics and clinical pharmacology, European Journal of Clinical Pharmacology 26 (1984) 663-668, doi:10.1007/BF00541922.
- Nature (London) 385, 303 (1997).
- M. Reist, P. A. Carrupt, E. Francotte, B. Testa: Chiral inversion and hydrolysis of thalidomide: mechanisms and catalysis by bases and serum albumin, and chiral stability of teratogenic metabolites. In: Chemical Research in Toxicology. 11, Nr. 12, 1998, S. 1521–8. doi:10.1021/tx9801817. PMID 9860497.
- Bernd Engels, Carsten Schmuck, Tanja Schirmeister, Reinhold Fink: Chemie für Mediziner. (google.de).
- Hermann J. Roth, Christa E. Müller und Gerd Folkers: Stereochemie & Arzneistoffe, Wissenschaftliche Verlagsgesellschaft Stuttgart, 1998, ISBN 3-8047-1485-4, S. 161–162.
- Duden: rectus.
- Hans-Dieter Jakubke, Hans Jeschkeit: Aminosäuren, Peptide, Proteine, Verlag Chemie, Weinheim, S. 30, 1982, ISBN 3-527-25892-2.
- H. J. Roth: Dex-, Lev-, Ar-, Es-, Rac-, neue „reine“ Arzneistoffe – Bilanz der letzten fünf Jahre. In: Deutsche Apothekerzeitung. Band 149, Nr. 28, 2009, S. 3182-6.
- H.J. Roth: Neue chirale Arzneistoffe – Eine Bilanz der Jahre 2009 bis 2013. In: Deutsche Apothekerzeitung. Nr. 4, 2014 (deutsche-apotheker-zeitung.de).
- H. J. Roth: Chirale versus achirale Arzneistoffe. In: Deutsche Apothekerzeitung. Nr. 6, 2019 (deutsche-apotheker-zeitung.de).
- H. J. Roth: Dex-, Lev-, Es-, eine Bilanz der letzten fünf Jahre: Trend zur Applikation reiner Enantiomere. In: Deutsche Apothekerzeitung. Band 144, Nr. 4, 2004, S. 2309.