Schwach koordinierende Ionen
Schwach koordinierende Ionen bezeichnen in der Chemie Ionen, die nur schwache Wechselwirkungen mit anderen Molekülen oder Ionen eingehen. Dabei werden die starken elektrostatischen Wechselwirkungen zwischen Kationen und Anionen durch eine Anzahl schwächerer Wechselwirkungen ersetzt. Diese Wechselwirkungen beziehen sich vor allem auf die Bildung von koordinativen Bindungen. Schwach koordinierende Ionen sind häufig große Moleküle und weisen Durchmesser im Nanometerbereich auf.
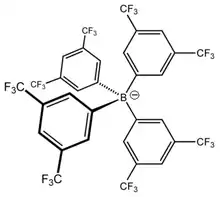
Die Löslichkeit von Salzen aus schwach koordinierenden Ionen in wenig oder nicht-polaren Lösungsmitteln ist höher als von klassischen Salzen; die reduzierte Neigung zur Ionenpaarungbildung kann in diesen Lösungsmitteln zu einer elektrischen Leitfähigkeit beitragen. Um zu betonen, dass die Ionen weitgehend unabhängig voneinander sind, werden außerdem die Begriffe freies Ion oder nacktes Ion verwendet. Solche Ionen sind in der Gasphase seit langem bekannt. Aus schwach koordinierenden Ionen werden zunehmend Verbindungen hergestellt, die vergleichbare Eigenschaften in Lösung oder im Festkörper aufweisen. Schwach koordinierende Ionen haben eine zunehmende Bedeutung, da sie die Untersuchung von hochreaktiven Verbindungen mit einer Vielzahl von physikalischen und chemischen Methoden ermöglichen. Praktische Anwendung finden schwach koordinierende Ionen beispielsweise bei der Herstellung von neuartigen Katalysatoren, in der koordinativen Polymerisation, bei der Entwicklung von ionischen Flüssigkeiten als Lösungsmittel für chemische Reaktionen und in der Elektrochemie.
Geschichte
In den 1970er Jahren galten komplexe Anionen wie BF4-, ClO4- oder Anionen hexahalogenierter Nicht- oder Halbmetalle der Stickstoffgruppe wie [SbCl6]- als nichtkoordinierende Anionen. Durch Kristallstrukturanalyse wurde jedoch erkannt, dass die in wässriger Lösung vorliegenden nichtkoordinierende Anionen im Festkörper durchaus koordinieren, wenn das Lösungsmittel entzogen wird.[1]
Um das Auftreten der Koordination von komplexen Anionen zu beschreiben, wurde der Begriff der "schwach koordinierenden Anionen" geprägt.[2] Diese galten als Ausgangspunkt für die Entwicklung immer schwächerer koordinierenden Anionen mit dem letztendlichen nicht zu erreichenden Ziel eines nichtkoordinierenden Anions.[3]
Ein Meilenstein wurde in den 1990er Jahren mit der Synthese des Tetrakis[3,5-bis(trifluormethyl)phenyl]borat-Ions ([B[3,5-(CF3)2C6H3]4]-) erreicht. Dieses Anion koordinierte weit schwächer als die bis dahin bekannten schwach koordinierenden Anionen und erlaubte das Studium stark elektrophiler Kationen.[4]
Grundlagen
Koordinative Bindung
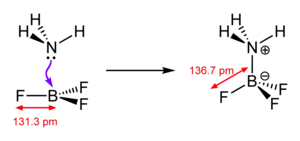
Eine koordinative Bindung bezeichnet eine chemische Bindung, bei der die Bindungselektronen nur von einem Bindungspartner bereitgestellt werden. Die bekanntesten Vertreter dieser Verbindungen sind ionische Komplexe. Dabei gruppieren sich mehrere negativ geladene Anionen um ein positiv geladenes Kation. Die Anionen nutzen ein freies Elektronenpaar, um als Ligand an das Kation, das Zentralatom, zu binden. Hierbei ist die Anzahl der umgebenden Ionen die Koordinationszahl und die räumliche Anordnung wird durch das Koordinationspolyeder dargestellt.
In festen Zustand sind Ionen in einem Ionengitter angeordnet. Dabei werden sowohl Kationen als auch Anionen von mehreren gegensätzlich geladenen Teilchen (den Gegenionen) umgeben.
Eine „schwache“ Koordination bedeutet in diesem Zusammenhang, dass die Bindungsenergie der koordinativen Bindung sehr gering ist. Da stets das Anion die Bindungselektronen beisteuert, ist die Koordinationsfähigkeit meist von der Beschaffenheit des Anions abhängig. Es ist, besonders in Festkörpern und Schmelzen, möglich, die Stärke der Bindung durch die Eigenschaften des Kations zu beeinflussen.
Freie Ionen in der Gasphase
Im Vakuum erzeugte Ionen gelten aufgrund der großen räumlichen Abstände zu jeglichen weiteren Atomen als frei im Raum schwebende Ladungsträger. Sie werden häufig in einer Ionenquelle durch gezielten Beschuss mit Elektronen (Stoßionisation) oder Ladungsübertragung durch ein anderes ionisiertes Gas (chemische Ionisation) erzeugt und hauptsächlich massenspektrometrisch untersucht.
In der Lebensmittelindustrie beispielsweise wird ionisierte Luft zur Pasteurisierung von Getränken verwendet. Hierbei wird die hohe Reaktivität der Ionen ausgenutzt. Dieser Umstand zeigt jedoch, dass solche Ionen meist nur eine sehr kurze Lebensdauer haben und praktisch direkt nach ihrer Erzeugung zerfallen oder weiter reagieren. Dadurch ist es nicht möglich, langwierige spektroskopische Untersuchungen (NMR, IR, Raman, UV/VIS) durchzuführen. Durch die Beschränkung auf die Gasphase sind Beugungsexperimente wie Röntgenbeugung oder Neutronenstreuung unmöglich.
Freie Ionen in Lösungen und Festkörpern
Die Definition von flüssigen und festen Aggregatzuständen bedingt, dass Teilchen stets untereinander wechselwirken. Daher kann es in diesen Zuständen keine „freien“ Ionen geben.
Einfluss des Lösungsmittels
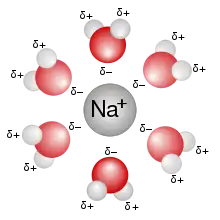
In Lösungen sind Ionen vom Lösungsmittel umgeben und solvatisiert. Das Lösungsmittel wirkt dabei als Dielektrikum (Isolator), indem es sich umgekehrt zur Ladung des Ions anordnet und so das elektrische Feld um das Ion abschwächt. Das Maß für diese Abschwächung ist die Polarität des Lösungsmittels, die sich durch seine Dielektrizitätskonstante (εr) ausdrückt.
In einem stark polaren Lösungsmittel wie etwa Wasser (εr = 80) zeigen gelöste Ionen kaum Wechselwirkung untereinander. Die Wechselwirkungen mit dem Lösungsmittel sind jedoch umso stärker, was sich am Beispiel von Lithium-Ionen veranschaulichen lässt: Aufgrund seiner großen Hydrathülle zeigt Li+ eine wesentlich geringere Beweglichkeit als die viel größeren Natrium- oder Kalium-Ionen.
Beim Übergang zu unpolaren Lösungsmitteln wie Dichlormethan (εr = 9) oder Diethylether (εr = 4,3) zeigt sich, dass viele Ionen stark aneinander koordinieren, was sich vor allem darin ausdrückt, dass die meisten Salze in solchen Lösungsmitteln unlöslich sind: Sie bilden starke Bindungen in Form eines Kristallgitters aus.
Ionen in Festkörpern
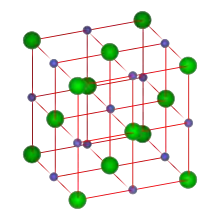
Im Festkörper ist das Maß für die Stärke der Wechselwirkungen zwischen den Ionen die Gitterenergie. Je größer dabei der Abstand von entgegengesetzt geladenen Ionen ist, umso kleiner wird die Gitterenergie. Dies lässt sich anhand der folgenden Tabelle demonstrieren:
| Name | Formel | Ionenradius der einwertigen Alkalimetall-Kationen X+ in pm |
Gitterenthalpie in kJ pro mol |
|---|---|---|---|
| Lithiumfluorid | LiF | 74 | 1039 |
| Natriumfluorid | NaF | 102 | 920 |
| Kaliumfluorid | KF | 138 | 816 |
| Rubidiumfluorid | RbF | 149 | 780 |
| Caesiumfluorid | CsF | 170 | 749 |
Beim Übergang von einatomigen zu mehratomigen Ionen verliert der Ionenradius an Aussagekraft, da nur die wenigsten Molekülionen vergleichbar hochsymmetrisch aufgebaut sind wie einzelne Atomionen. Besonders bei eher kleinen, aber sehr unsymmetrischen Ionen weichen die mit Hilfe der Ionenradien berechneten Gitterenergien weit von den experimentellen Werten ab. Um diesem Umstand Rechnung zu tragen, wurde von Donald Jenkins das Konzept des Thermochemischen Volumens eingeführt.[5]
Hierbei wird das Volumen eines Ions aus dem Volumen der Elementarzelle eines Ionenkristalls berechnet und daraus die Gitterenergie ermittelt. Als semiempirische Methode stimmen die berechneten Gitterenergien in vielen Fällen mit den experimentellen Werten überein.[5]
Konzepte
Der Ansatz zur Entwicklung schwach koordinierender Ionen besteht darin, eine geringe Ladung über ein möglichst großes Volumen zu verteilen. Dadurch wird die Gitterenergie im Festkörper (und damit die Wechselwirkung zwischen entgegengesetzt geladenen Ionen) minimiert. Darüber hinaus muss das Ion eine geringe Polarisierbarkeit aufweisen, damit ein in der Nähe befindliches Gegenion oder Lösungsmittelteilchen keine Ladungsschwerpunkte erzeugen kann. Diese würden sonst wiederum Dipol-Dipol-Kräfte bewirken und so zu einer Koordination führen.
Die leichte Polarisierbarkeit ist der Grund, warum große einatomige Ionen wie etwa Iodid oder Caesium nur begrenzt als schwach koordinierende Ionen wirken. Heutzutage konzentriert sich die Forschung daher auf die Herstellung von sehr großen einwertigen (einfach positiv oder negativ geladenen) Molekülen.
Schwach koordinierende Anionen
Schwach koordinierenden Anionen werden hauptsächlich nach zwei unterschiedlichen Methoden verwirklicht.
Kovalent gebundene Gerüstanionen
Eine Möglichkeit ist der Aufbau eines vielatomigen, negativ geladenen Gerüsts, das eine möglichst kugelförmige Oberfläche besitzt, auf der die Ladung verteilt wird. Die Atome des Gerüsts werden durch starke kovalente Bindungen zusammengehalten.
Hauptvertreter dieser Klasse sind negativ geladene Carborane wie etwa [CB11H12]−. Durch die Substitution aller H-Atome konnte das noch stabilere Carborat [1-R-CB11F11]− (R = Me, Et) erhalten werden, das als bislang bestes schwach koordinierendes Anion gehandelt wird.[6]
Stabile Lewis-Säure-Base-Komplexe
Die zweite Herangehensweise ist der Aufbau von besonders stabilen Komplexanionen aus starken Lewis-Säuren und Lewis-Basen. Aus einem Kation mit der Ladung X sowie X+1 negativ geladenen Liganden entsteht ein Komplex mit einer Gesamtladung von −1. Wichtig für die Stabilität des Komplexes ist eine starke koordinative Bindung der Liganden an das Zentralatom. Bislang werden hierfür hochgeladene Kationen wie B3+, Al3+, As5+, Sb5+, Nb5+, Y3+ oder La3+ eingesetzt.
Für die Ausbildung einer starken Bindung eignen sich als Liganden besonders Atome mit einer hohen Elektronegativität wie Fluor oder Sauerstoff. Anionen wie Tetrafluoroborat ([BF4]−), Perchlorat ([ClO4]−) oder Hexafluoroantimonat ([SbF6]−) werden bereits vielfach in der Industrie verwendet. Es ist jedoch erwiesen, dass solche Anionen in unpolaren Lösungsmitteln vergleichsweise stark an Kationen koordinieren.[7]
| Häufig eingesetzte Liganden | ||
|---|---|---|
| Bezeichnung | Summenformel | perfluoriert |
| Alkyl- | ||
| Methyl- | -CH3 | -CF3 |
| t-Butyl- | -C(CH3)3 | -C(CF3)3 |
| Aryl- | ||
| Phenyl- | -C6H5 | -C6F5 |
| Alkoxy- | ||
| Methoxy- | -O-CH3 | -O-CF3 |
| t-Butoxy- | -O-C(CH3)3 | -O-C(CF3)3 |
| Aryloxi- | ||
| Phenyloxi- | -O-C6H5 | -O-C6F5 |
| Perfluorotelluroxi- | ||
| Teflat | -O-TeF5 | |
In der Forschung werden daher zunehmend Liganden eingesetzt, die über voluminöse Substituenten und eine chemisch inerte Oberfläche verfügen. Bedeutende Vertreter sind Alkyl- und Arylliganden wie das [BPh4]−(Kalignost). Von den entsprechenden Alkoholen abgeleitet ergeben sich die Alkoxi- und Aryloxiliganden, die über den Sauerstoff an das Zentralatom gebunden sind.
Um die Oberfläche der Ionen chemisch unangreifbar (inert) zu machen, werden perfluorierte Varianten der Liganden eingesetzt (siehe Fluorcarbone und Fluorkohlenwasserstoffe). So ist beispielsweise das mit perfluorierten tert-Butanolliganden gebildete Anion [Al[OC(CF3)3]4]− in seinen Eigenschaften vergleichbar mit dem industriell häufig eingesetzten [SbF6]−.
- Beispiele
4)-.jpg.webp) [B(CF3)4]−
[B(CF3)4]−4)-.jpg.webp) [B(C6F5)4]−
[B(C6F5)4]−4)-.jpg.webp) [Al{OC(CF3)3}4]−
[Al{OC(CF3)3}4]−
Schwach koordinierende Kationen
Zur Herstellung von schwach koordinierenden Kationen gibt es bislang kaum übergeordnete Konzepte. So konzentriert sich die Entwicklung zumeist auf den angestrebten Verwendungszweck, beispielsweise die Stabilisierung von „nackten“ Fluoridionen und anderen hochreaktiven Anionen.
Gemeinsamkeiten von schwach koordinierenden Kationen ist häufig der Aufbau von voluminösen Molekülen mit einem positiv geladenen Stickstoff, Phosphor oder Schwefel. Der Rest des Moleküls ist dabei so konstruiert, dass sie nur sehr schwache Brønsted-Säuren darstellen, also keine Protonen abspalten können, was andernfalls zur Zersetzung des Kations führen würde. Damit können diese Moleküle als Salze von sehr starken Basen angesehen werden, eine Eigenschaft, die weitere Anwendungsmöglichkeiten eröffnet.
Die folgende Tabelle listet einige Kationen auf, bei denen schwach koordinierte Fluoridverbindungen nachgewiesen sind:
| Bezeichnung | Summenformel | Strukturformel |
|---|---|---|
| Tetramethylammonium[8] | [N(CH3)4]+ | 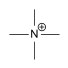 |
| Tetramethylphosphonium[9] | [P(CH3)4]+ |  |
| Tetrakis(dimethylamino)phosphonium[10] | {P[N(CH3)2]4}+ | 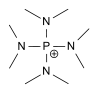 |
| Tris(dimethylamino)sulfonium[11] | {S[N(CH3)2]3}+ | 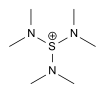 |
| Hexamethylpiperidinium[12] | [C11H24N]+ | 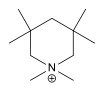 |
| Hexakis(dimethylamino)diphosphazenium Schwesingerbase P2[13] |
{[(NMe2)3]P=N=P[(NMe2)3]}+ | 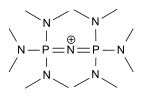 |
Thermodynamische Eigenschaften
Durch Messungen und Berechnungen unter Zuhilfenahme des Born-Haber-Kreisprozesses ist es möglich, thermodynamische Eigenschaften von Verbindungen mit großen und schwach koordinierenden Ionen zu bestimmen. Der Vergleich der Eigenschaften für unterschiedliche Ionen ist ein Maß für die Güte eines bestimmten Ions.
Die Berechnung der Gitterenergie von festen Verbindungen mit sehr großen schwach koordinierenden Anionen, liefert – abhängig vom Volumen der Teilchen – sehr kleine Werte.
| Salz | thermochemisches Volumen in Å3 | Gitterenergie Upot. in kJ mol−1 |
|---|---|---|
| Li+F− | 27 | 1036 |
| Cs+F− | 43 | 740 |
| Cs+[AsF6]− | 128 | 568 |
| Cs+[Al{OC(CF3)4}]− | 776 | 362 |
| [Ag(S8)2]+[Al{OC(CF3)4}]− | 1169 | 326 |
Diese Werte lassen sich gut mit den Sublimationsenthalpien von sehr schweren Molekülen (z. B. den Fullerenen C60 und C70) vergleichen:
| Salz | Molmasse in g mol−1 | Energie in kJ mol−1 |
|---|---|---|
| [Ag(S8)2]+[Al{OC(CF3)4}]− | 1588 | 326 |
| C70 | 841 | 200 |
| C60 | 721 | 175 |
Die schwach koordinierten Verbindungen haben im Festkörper demnach Energien, die mit Molekülen in der Gasphase vergleichbar sind. Tatsächlich war das oben genannte Kation [Ag(S8)2]+ vor der Darstellung mit einem schwach koordinierenden Anion nicht bekannt. Durch die grundsätzlich niedrige Gitterenergie sind destabilisierende Effekte, wie sie durch sehr schwach gebundene Liganden (wie in diesem Fall Schwefel) auftreten, in ihrer Wirkung vermindert. Umgekehrt kann der Einsatz von schwach koordinierenden Ionen solche Komplexe stabilisieren.
Vergleichbare Bedingungen können ansonsten teilweise durch Isolation der Ionen in einer Matrix oder mit Hilfe von geringfügig modifizierten Zeolithen erreicht werden.
Aufgrund dieser niedrigen Gitterenergien ergeben sich eine Reihe weiterer Eigenschaften. Solche Salze sind viel eher in unpolaren Lösungsmitteln mit einer niedrigen Dielektrizitätskonstante löslich, da die Energiebarriere zur Lösung der Ionenbindung und Solvatation durch das Lösungsmittel geringer ist als bei herkömmlichen Salzen. Darüber hinaus wird die Gitterenergie in einigen Verbindungen bereits bei Raumtemperatur überschritten, so dass einige Salze schon bei diesen niedrigen Temperaturen flüssig sind; Kochsalz hingegen schmilzt erst bei Temperaturen über 800 °C.
Anwendungen
Die Anwendungsmöglichkeiten sind sehr vielfältig, in diesem Abschnitt werden daher nur einige Beispiele genannt.
Koordinationspolymerisation
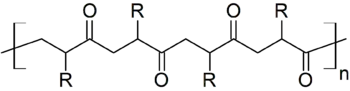
Die kationische oder anionische Koordinationspolymerisation ist eine der wichtigsten Verfahren zur Polymerisation von Alkenen. Häufig werden hierfür positiv geladene Metallocene mit Gruppe-IV Elementen wie Titan oder Zirconium verwendet. Die Aktivität des Katalysators hängt jedoch stark von den Eigenschaften des Gegenions ab. Erst bei Einsatz schwach koordinierender Ionen wie [B(C6F5)4]− haben die gebildeten Salze die erforderliche hohe Aktivität. Diese Steigerung der Reaktivität wird nachweislich dem verringerten Einfluss des Anions zugeschrieben.[14]
Zur Copolymerisation von Kohlenstoffmonoxid mit Ethylen oder Propylen kann ein kationischer Palladium(II)-Katalysator mit einem Tetrakis[3,5-bis (trifluormethyl)phenyl]borat-Anion verwendet werden. Durch die Reaktion entstehen Polyketone.[15]
Lithium-Ionen-Akkus
Eine der bekanntesten Anwendungsgebiete innerhalb der Elektrochemie sind die Lithium-Ionen-Akkus. Um eine hohe Batteriespannung zu erreichen, ist die Wahl des Gegenions und des Lösungsmittels entscheidend. Je schwächer das Gegenion und je unpolarer das gewählte Lösungsmittel, umso höher ist die Ionenbeweglichkeit der Lithiumionen und damit die Leitfähigkeit des Lithiumelektrolyten. Gegenwärtig wird in diesem Bereich am häufigsten Li+[PF6]− eingesetzt, es gibt jedoch bereits eine Reihe von Patentanmeldungen für den Einsatz anderer Anionen.
Ionische Flüssigkeiten
Durch ihre besonderen Eigenschaften eröffnen Ionische Flüssigkeiten viele Anwendungsmöglichkeiten als Lösungsmittel in der organischen Chemie, als Elektrolyte oder in der Katalyse.[16] Beruhend auf den sehr niedrigen Schmelzpunkten sowie äußerst niedrigen Dampfdrücken, unterschiedlichen Löslichkeiten in polaren und unpolaren Lösungsmitteln und schließlich aufgrund ihrer Umweltverträglichkeit ist zu erwarten, dass ionische Flüssigkeiten viele organische Lösungsmittel verdrängen werden.
Organische Katalyse
In der katalytischen organischen Chemie werden mitunter hochreaktive Kationen wie Ag+, Li+ oder auch F− eingesetzt. Der Einsatz von schwach koordinierenden Ionen ermöglicht dabei die Reaktionen bei niedrigeren Temperaturen ablaufen zu lassen. So sind Fluorierungsreaktionen von Aromaten, bei denen beispielsweise Chlorbenzol direkt in Fluorbenzol umgewandelt wird, nur mit sehr reaktiven Fluoridionen möglich.
Literatur
- Ingo Krossing, Ines Raabe: Nichtkoordinierende Anionen – Traum oder Wirklichkeit? Eine Übersicht zu möglichen Kandidaten. In: Angewandte Chemie. 116, 2004, S. 2116–2142, doi:10.1002/ange.200300620.
- A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 101. Auflage. Walter de Gruyter, Berlin 1995, ISBN 3-11-012641-9.
- E. Riedel (Hrsg.); Christoph Janiak, T. M. Klapötke, H.-J. Meyer: Moderne anorganische Chemie. Berlin 2007, ISBN 978-3-11-019060-1 (Grundlagen zur Koordinationschemie)
Weblinks
Einzelnachweise
- Michael R. Rosenthal: The myth of the non-coordinating anion. In: Journal of Chemical Education. 50, 1973, S. 331, doi:10.1021/ed050p331.
- Ingo Krossing, Ines Raabe: Nichtkoordinierende Anionen – Traum oder Wirklichkeit? Eine Übersicht zu möglichen Kandidaten. In: Angewandte Chemie. 116, 2004, S. 2116–2142, doi:10.1002/ange.200300620.
- Steven H. Strauss: The search for larger and more weakly coordinating anions. In: Chemical Reviews. 93, 1993, S. 927–942, doi:10.1021/cr00019a005.
- Neal A. Yakelis, Robert G. Bergman: Safe Preparation and Purification of Sodium Tetrakis[(3,5-trifluoromethyl)phenyl]borate (NaBArF24): Reliable and Sensitive Analysis of Water in Solutions of Fluorinated Tetraarylborates. In: Organometallics. 24, 2005, S. 3579–3582, doi:10.1021/om0501428.
- H. Donald B. Jenkins, Helen K. Roobottom, Jack Passmore, Leslie Glasser: Relationships among Ionic Lattice Energies, Molecular (Formula Unit) Volumes, and Thermochemical Radii. In: Inorganic Chemistry. 38, 1999, S. 3609–3620, doi:10.1021/ic9812961.
- Daniel Stasko, Christopher A. Reed: Optimizing the Least Nucleophilic Anion. A New, Strong Methyl Reagent. In: Journal of the American Chemical Society. 124, 2002, S. 1148–1149, doi:10.1021/ja0118800.
- Wolfgang Beck, Karlheinz Suenkel: Metal complexes of weakly coordinating anions. Precursors of strong cationic organometallic Lewis acids. In: Chemical Reviews. 88, 1988, S. 1405–1421, doi:10.1021/cr00089a017.
- Karl O. Christe, William W. Wilson: Reaction of the fluoride anion with acetonitrile. Chloroform and methylene chloride. In: Journal of Fluorine Chemistry. 47, 1990, S. 117–120, doi:10.1016/S0022-1139(00)80453-4.
- Andreas Kornath, F. Neumann, H. Oberhammer: Tetramethylphosphonium Fluoride: “Naked” Fluoride and Phosphorane. In: Inorganic Chemistry. 42, 2003, S. 2894–2901, doi:10.1021/ic020663c.
- Alexander Kolomeitsev, Valery Movchun, Eduard Rusanov, German Bissky, Enno Lork, Gerd-Volker Röschenthaler, Peer Kirsch: Different fluoride anion sources and (trifluoromethyl)trimethylsilane: molecular structure of tris(dimethylamino)sulfonium bis(trifluoromethyl)trimethylsiliconate, the first isolated pentacoordinate silicon species with five Si–C bonds. In: Chemical Communications. S. 1017–1018, doi:10.1039/A901953G.
- Andrew E. Bayliff, Richard D. Chambers: Reactions involving fluoride ion. Part 34. Stable perfluorinated carbanions. In: Journal of the Chemical Society, Perkin Transactions 1. 1988, S. 201–208, doi:10.1039/P19880000201.
- Ali Reza Mahjoub, Xiongzhi Zhang, Konrad Seppelt: Reactions of the “Naked” Fluoride Ion: Syntheses and Structures of SeF62− and BrF6−. In: Chemistry – A European Journal. 1, 1995, S. 261–265, doi:10.1002/chem.19950010410.
- Reinhard Schwesinger, Reinhard Link, Gerhard Thiele, Heinz Rotter, Dieter Honert, Hans-Heinrich Limbach, Ferdinand Männle: Stabile Phosphazenium-Ionen in der Synthese – ein leicht zugängliches, extrem reaktives „nacktes“ Fluoridsalz. In: Angewandte Chemie. 103, 1991, S. 1376–1378, doi:10.1002/ange.19911031031.
- Eugene You-Xian Chen, Tobin J. Marks: Cocatalysts for Metal-Catalyzed Olefin Polymerization: Activators, Activation Processes, and Structure-Activity Relationships. In: Chemical Reviews. 100, 2000, S. 1391–1434, doi:10.1021/cr980462j.
- M. Brookhart, Francis C. Rix, J. M. DeSimone, James C. Barborak: Palladium(II) catalysts for living alternating copolymerization of olefins and carbon monoxide. In: Journal of the American Chemical Society. 114, 1992, S. 5894–5895, doi:10.1021/ja00040a082.
- Jeffrey A. Boon, Joseph A. Levisky, J. Lloyd Pflug, John S. Wilkes: Friedel-Crafts reactions in ambient-temperature molten salts. In: The Journal of Organic Chemistry. 51, 1986, S. 480–483, doi:10.1021/jo00354a013.