Bergius-Pier-Verfahren
Das Bergius-Pier-Verfahren (auch als Bergius-Verfahren oder I.G.-Verfahren bezeichnet) ist ein großtechnisches Verfahren zur Kohleverflüssigung durch direkte Hydrierung von Kohle. Dabei werden die Makromoleküle der Kohle durch Wasserstoff bei hohen Drücken und Temperaturen zu kleineren Molekülen abgebaut. Die Rohstoffbasis bestand anfangs aus Braunkohle, die durch Kohlevergasung sowohl den zur Hydrierung notwendigen Wasserstoff lieferte als auch die Kohlenstoffbasis für die Produkte bildete. Später erweiterten Steinkohle, Teere und Teeröle die Rohstoffpalette.
_Chemist_of_Heidelberg_Wellcome_L0003155.jpg.webp)
Als Produkte entstehen gasförmige und flüssige Kohlenwasserstoffe, die als Kraft- und Schmierstoffe dienen. In der Zeit zwischen 1910 und 1925 entwickelte der deutsche Chemiker Friedrich Bergius die Grundlagen des Prozesses. Die kommerzielle Verfahrens- und Katalysatorentwicklung erfolgte ab 1925 durch die BASF unter der Projektleitung von Matthias Pier.
Die I.G. Farben baute 1927 in Leuna die erste Hydrieranlage nach dem Bergius-Pier-Verfahren, ab Mitte der 1930er Jahre ordnete das Deutsche Reich im Rahmen der Autarkiebestrebungen den Auf- und Ausbau von weiteren Hydrierwerken an. Ab Mitte der 1930er Jahre waren die Anlagen im Deutschen Reich im Rahmen der Autarkiebestrebungen von besonderer Bedeutung. Während des Zweiten Weltkrieges deckte das Bergius-Pier-Verfahren einen Großteil des Kraftstoffbedarfs der deutschen Wehrmacht.
In der Nachkriegszeit verdrängten preiswerte Produkte auf Basis von Erdöl die kohlestämmigen Erzeugnisse, der Betrieb der Hydrierwerke wurde eingestellt. Engpässe in der Erdölversorgung während der Ölkrise in den 1970er Jahren verstärkten das Interesse an dem Verfahren wieder. Eine großtechnische Umsetzung erfolgte jedoch in der westlichen Welt nicht. Im Gegensatz dazu begann das an Kohlevorräten reiche China 2003 den Bau eines Hydrierwerks in der Inneren Mongolei, das Kraftstoffe für den chinesischen Markt produziert.
Die Nobelstiftung vergab für die Entwicklung des Verfahrens 1931 den Nobelpreis für Chemie an Friedrich Bergius zusammen mit Carl Bosch „für ihre Verdienste um die Entdeckung und Entwicklung der chemischen Hochdruckverfahren“.
Geschichte

Im Jahr 1869 führte der französische Chemiker Marcelin Berthelot erste Versuche zur Gewinnung von flüssigen Kohlenwasserstoffen aus Kohle durch. Dazu ließ er einen Iodwasserstoffüberschuss bei Temperaturen von 270 °C auf Kohle einwirken.[1] Iodwasserstoff zerfällt bei dieser Temperatur in die Elemente und lieferte den Wasserstoff für die Kohlehydrierung. Die Reaktion führte zur Bildung flüssiger Kohlenwasserstoffe, erlangte jedoch keine technische Bedeutung. Berthelots Versuche initiierten dem ungeachtet weitere Forschungen auf dem Gebiet der Kohlehydrierung. Um 1900 etablierte Paul Sabatier die katalytische Hydrierung als neues chemisches Verfahren, für das er 1912 mit dem Nobelpreis in Chemie ausgezeichnet wurde. Die Erkenntnis, dass Metalle wie Nickel oder Eisen die Hydrierung organischer Stoffe katalysieren, sowie die Arbeiten von Wladimir Nikolajewitsch Ipatjew über Hochdruckhydrierungen, bildeten eine wichtige Grundlage für Bergius Arbeiten.[2]
In Deutschland gründete die Kaiser-Wilhelm-Gesellschaft unter anderem zu diesem Zweck das Kaiser-Wilhelm-Institut für Kohlenforschung.[3]
„Wie schön wäre es nun, wenn man aus den festen Brennmaterialien durch einen passenden Reduktionsprozess auf ökonomische Weise flüssige Brennstoffe herstellen könnte!“
Mit diesen Worten beschrieb der Nobelpreisträger Emil Fischer 1912 anlässlich der Gründung dieses Instituts seinen Wunsch, ein „fundamentales Problem der Heizstoffindustrie“ zu lösen, besonders in Bezug auf Deutschland und dessen Mangel an flüssigen fossilen Brennstoffen. Den von Fischer gewünschten Reduktionsprozess, der in die chemische Technik überführt werden konnte, entwickelte schließlich Friedrich Bergius.[4]
Anfänge in Hannover und Essen
Als Privatdozent für Reine und Angewandte Physikalische Chemie an der Königlich-technischen Hochschule Hannover, wo er über den Inkohlungsprozess habilitiert hatte, führte Bergius schon 1910 Hydrierversuche an synthetischer Kohle durch.[5] Bereits erste Tests führten zu reproduzierbaren Ergebnissen, die bei Temperaturen von 400 bis 450 °C und einem Wasserstoffdruck von 150 bar in 80%iger Ausbeute gasförmige und flüssige Kohlenwasserstoffe lieferten.[5] Bergius erhielt 1913 ein Patent für seine Erfindung.[6]
Die Überführung des Verfahrens vom Labor in eine Großanlage erwies sich als schwierig und erforderte erhebliche finanzielle Mittel. Durch Kontakte zu Karl Goldschmidt trat Bergius in die Leitung der Chemischen Fabrik Goldschmidt in Essen ein, welche die Verfahrensentwicklung zunächst finanzierte. In der Zeit nach dem Ersten Weltkrieg fehlten jedoch die finanziellen Mittel für die Entwicklung eines geeigneten Katalysators, was in der Folge zur Beendigung der Zusammenarbeit führte.[5]
Gründung der I.G. Farben

Der steigende Motorisierungsgrad sowie die beginnende Umstellung der Schifffahrt und des Schienenverkehrs auf flüssige Kraftstoffe ließen die Gewinnung von Kraft- und Schmierstoffen durch die Kohlehydrierung als eine gewinnträchtige Einnahmequelle erscheinen.[7] Die Anhörungen vor dem Federal Oil Conservation Board zeigten im Jahr 1926, dass man sich schon damals um die Erschöpfung der Erdölreserven Sorge machte.[8]:6 Die im Gegensatz dazu reichen Braun- und Steinkohlevorkommen dagegen schienen auf Jahrhunderte die Versorgung mit Kraftstoffen sichern zu können.[9]
Durch die technischen und kommerziellen Erfolge der Hochdruckchemie beim Haber-Bosch-Verfahren, der Methanolherstellung und der Isobutylölsynthese ermutigt, lenkte Carl Bosch daher die Forschungsaktivitäten der BASF auf die Hochdruckhydrierung der Braunkohle.[10] Im Juli 1925 erwarb die BASF einen Teil der deutschen Bergius-Patente und ein Jahr später den größten Teil der internationalen Rechte von der Royal Dutch Shell.[11] Die Verfahrensentwicklung übertrug die BASF dem Nernst-Schüler Matthias Pier, der schon das Hochdruckverfahren für die Herstellung von Methanol entwickelt hatte. Er führte in den Ammoniak- und Methanolapparaturen Versuche zur katalytischen Hydrierung von Braunkohlenteer mit schwefelhaltigen Katalysatoren wie Eisen- und Molybdänsulfid durch. Carl Krauch, der spätere Leiter des Reichsamts für Wirtschaftsausbau, förderte Piers Arbeiten.
Die Hydrierung von Kohle in einer Suspension stellte gegenüber den reinen Gasphasenreaktionen eine Herausforderung dar und erforderte erhebliche Investitionen in die Verfahrensentwicklung. Eine hohe Konzentration von typischen Katalysatorgiften erforderte eine hohe Stabilität und Selektivität des Katalysators, um die Reaktionen bei einem sehr inhomogenen Rohstoff zu den gewünschten Produkten zu führen. Dazu kamen technische Schwierigkeiten wie der Angriff von Wasserstoff und Schwefelwasserstoff auf das Reaktormaterial sowie Probleme bei der Trennung von Restkohle, Asche und Katalysator von den verflüssigten Produkten.[12]
Carl Bosch erkannte, dass die Beschaffung des benötigten Kapitals für die Überführung des Verfahrens in die chemische Technik eine breite finanzielle Basis benötigte, was 1925 letztlich zur Gründung der I.G. Farben führte.[13] Wegen der Nähe zum Mitteldeutschen Braunkohlerevier und den da vorhandenen Erfahrungen in der Hochdrucktechnik wählte Bosch die Leunawerke 1927 als erste Produktionsstätte aus. Innerhalb von fünf Jahren überwand die BASF die technischen Schwierigkeiten und steigerte den Ausstoß von Kraftstoffen auf etwa 100.000 Tonnen pro Jahr. Gegenüber den Produkten der indirekten Kohlehydrierung durch die Fischer-Tropsch-Synthese wies das nach dem Bergius-Pier-Verfahren produzierte synthetische Benzin eine höhere Oktanzahl auf.
Die I.G. Farben investierte bis 1931 etwa 426 Millionen Reichsmark in das Verfahren, mehr als das Doppelte des Umsatzes des BASF-Teils der I.G. Farben in diesem Jahr.[14] Die Standard Oil of New Jersey erwarb in dieser Zeit die Lizenz zur weltweiten Vermarktung des Verfahrens.[15] Die Firmen Imperial Chemical Industries (ICI), Shell, Standard Oil und I.G. Farben gründeten 1931 die International Hydrogenation Patents Company, um die Ressourcen und das Know-how für die Herstellung synthetischen Benzins zu bündeln und um die Veröffentlichung von Entdeckungen einzuschränken.[16]
Größer als die technischen waren die wirtschaftlichen Probleme. Die Herstellungskosten des Leuna-Benzins erwiesen sich teilweise höher als die für die Benzinherstellung auf der Basis von Erdöl. Zeitweise lag der Preis für Benzin aus Erdöl unter dem Preis der eingesetzten Kohle.[12] Zudem erwies sich die pessimistische Prognose bezüglich der gewinnbaren Erdölvorkommen als falsch. Die einsetzende Weltwirtschaftskrise erhöhte die Nettoverluste der I.G. Farben, die auch nicht durch die Zahlungen von Lizenzgebühren der Standard Oil of New Jersey auszugleichen waren.[15]
Zeit des Nationalsozialismus

1933 nahmen Vertreter der I.G. Farben Kontakte zur nationalsozialistischen Führung auf, um für eine politische Unterstützung des Verfahrens zu werben. Das Reichsfinanzministerium gewährte daraufhin Schutzzölle, um das synthetische Benzin konkurrenzfähig zu halten. Es kam im Dezember 1933 zur Unterzeichnung des vom wirtschaftspolitischen Sprecher der NSDAP Gottfried Feder und Carl Bosch ausgehandelten Feder-Bosch-Abkommens, wobei sich die I.G. Farben zur Lieferung von synthetischem Benzin zu einem garantierten Preis verpflichtete.[17]
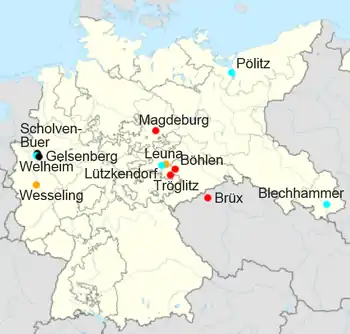
Hitler befahl 1936 in der von ihm verfassten Denkschrift zum Vierjahresplan die Durchführung eines Vierjahresplans, unter anderem mit dem Ziel des maximalen Ausbaus von Anlagen zur Herstellung synthetischen Benzins.[18] Eine wesentliche Rolle sollte hierbei die Braunkohle-Benzin AG (Brabag) mit ihren vier Hydrierwerken in Magdeburg, Böhlen, Schwarzheide und Zeitz einnehmen. Insgesamt entstanden im Deutschen Reich nach der Fischer-Tropsch-Synthese der Ruhrchemie neun Werke und zwölf nach dem Hochdruck-Hydrierungsverfahren der I.G. Farben.
Die größten Anlagen mit einer Produktionskapazität von über 600.000 t/a (Tonnen Treibstoffe pro Jahr) arbeiteten in Pölitz, Leuna und Brüx.[5][19] Die zwölf Bergius-Pier-Hydrierwerke hatten zusammen eine geplante Kapazität von 3.866.000 t/a und die neun Fischer-Tropsch-Anlagen von 647.000 t/a. Die geplante Jahreskapazität wurde jedoch bei allen Werken erst 1943/44 erreicht.[20]
| Produktionsangaben von deutschen Hydrierwerken nach dem Bergius-Pier-Verfahren[21] | ||||
|---|---|---|---|---|
| Produktions- beginn |
Standort | Betreiber | Einsatzstoff | Produktionskapazität 1943/44 in Tonnen pro Jahr |
| 1927 | Leuna | I.G. Farben | Braunkohle | 650.000 |
| 1936 | Böhlen | BRABAG | Braunkohlenteer | 250.000 |
| 1936 | Magdeburg | BRABAG | Braunkohlenteer | 220.000 |
| 1936 | Scholven-Buer | Hibernia AG | Steinkohle | 280.000 |
| 1937 | Bottrop-Welheim | Ruhröl GmbH | Pech | 130.000 |
| 1939 | Gelsenberg | Gelsenkirchen-Benzin AG | Steinkohle | 400.000 |
| 1939 | Tröglitz/Zeitz | BRABAG | Braunkohlenteer | 280.000 |
| 1940 | Lützkendorf | Mitteldeutsche Treibstoff- und Oelwerke A.G. | Teer, Öl | 50.000 |
| 1940 | Pölitz | Hydrierwerke Pölitz AG | Steinkohle, Öl | 700.000 |
| 1941 | Wesseling | Union Rheinische Braunkohlen Kraftstoff AG | Braunkohle | 260.000 |
| 1943 | Brüx | Nordböhmische Kohlenwerks-AG | Braunkohlenteer | 600.000 |
| 1943 | Blechhammer | Oberschlesische Hydrierwerke AG | Steinkohle, Teer | 420.000 |
| bis 1944 | Bergius-Pier gesamt | 4.230.000 | ||
Die gemeinsam mit den Fischer-Tropsch-Anlagen einmalig im Zeitraum von 1943 bis Anfang Mai 1944 erreichte Höchstleistung von zusammen rund 4,5 Millionen Tonnen ist jedoch nicht mit der Jahresproduktion synthetisch hergestellten Flugbenzins gleichzusetzen, was in der Nachkriegszeit verschiedene Autoren angaben. Den größten Produktanteil des I.G.-Verfahrens bildeten zwar Flugtreibstoffe, jedoch gefolgt von Fahrbenzin und Dieselkraftstoff, Heizöl, Schmieröl und sonstigen Produkten (Sekundärprodukte). Mit der Fischer-Tropsch-Synthese wurde zu dieser Zeit weder Flugtreibstoff noch Heizöl hergestellt, so dass der Kraftstoffanteil, der durch dieses Verfahren erzeugt wurde, geringer war als bei dem I.G.-Verfahren. Fahrbenzin und Dieselöl konnten mit beiden Verfahren produziert werden, wobei sich die Qualität der Produkte durch verschiedene Oktanzahlen beziehungsweise Cetanzahlen unterschieden. Konkret betrug die Produktion aller 21 Hydrierwerke im Jahr 1943 gerundet 3 Millionen Tonnen Benzin (Fahr- und Flugbenzin zusammen), 770.000 Tonnen Schmieröl und 430.000 Tonnen Dieselkraftstoff.[22][23]
Zweiter Weltkrieg

Obwohl die Versorgung mit Kraft- und Schmierstoffen eine der anfälligsten Stellen der deutschen Kriegsmaschinerie war, erfolgte ein direkter Angriff auf die Hydrierwerke erst ab 1944.[24] Nach den Erfolgen der Operation Tidal Wave bei den Luftangriffen auf Ploiești und die dortigen Raffinerien und Erdölreserven erklärte die Führung der alliierten Luftwaffe die deutschen Hydrierwerke zum strategischen Angriffsziel. Am Abend des 12. Mai 1944 starteten 935 Bomber der United States Army Air Forces einen Großangriff auf die Hydrierwerke Leuna, Böhlen, Tröglitz, Lützkendorf und Brüx. Die im Zuge dieser Luftangriffe erreichten Zerstörungen legten den Betrieb der fünf Hydrierwerke vorübergehend still.[25]
Bis zum 29. Mai 1944 flogen die US-Bomberverbände konzentrierte Angriffe gegen alle 27 im deutschen Einflussbereich liegenden Raffinerie- und Hydrieranlagen. Die Werke waren danach erheblich, meist total zerstört. Damit drohte der vollständige Zusammenbruch der deutschen Treibstoffversorgung. Vor diesem Hintergrund ordnete Adolf Hitler umgehend und persönlich am 30. Mai 1944 „Sofortmaßnahmen zur Wiederinbetriebnahme und zum Schutz der Hydrierwerke“ an.[26] Mit dem geheim gehaltenen Mineralölsicherungsplan wurde innerhalb kürzester Zeit versucht, die Werke wieder aufzubauen und die Anlagen durch den Einsatz massiver Flak in „Hydrierfestungen“ zu verwandeln.[27]
Die Instandsetzung gelang infolge fortwährender Luftangriffe der Alliierten lediglich temporär. Ab September 1944 konnten sämtliche deutschen Hydrierwerke der Wehrmacht monatlich zusammen nur noch 11.000 Tonnen Benzin liefern.[28] Auch das von Rüstungsminister Albert Speer initiierte Projekt, Hydrierwerke unter Einsatz von Tausenden von Fachkräften, Zwangsarbeitern und Strafgefangenen unter Tage in Stollen zu verlagern, blieb erfolglos. Die Fertigstellung gelang vor Kriegsende nicht mehr.[29] Schließlich trug die Zerstörung der deutschen Hydrieranlagen dazu bei, dass die deutsche Kriegsmaschinerie zum Erliegen kam.[24]
Andere Länder
Die Möglichkeiten der Umwandlung fossiler Brennstoffe in flüssige Kraft- und Schmierstoffe förderten andere Länder, insbesondere Großbritannien und die USA, zumindest in der Anfangszeit vergleichbar. Ausschlaggebend dafür war eine von US-Wissenschaftlern im Jahr 1925 veröffentlichte und weltweit ernstgenommene These, wonach die globalen Erdölvorräte in sieben Jahren erschöpft seien, während die Motorisierung unaufhaltsam zunehme. Ein Jahr später gründete der Kongress der Vereinigten Staaten ein Komitee mit der Aufgabe, nach Alternativen zu suchen.[30][31]
In der Folge erlangten, ähnlich der I.G. Farben in Deutschland, die Imperial Chemical Industries (ICI) in Großbritannien und DuPont in den USA eine nationalökonomische, politische Bedeutung. Zur Herstellung synthetischen Benzins entstanden in Zusammenarbeit mit der I.G. Farben ab 1927 in den USA zwei Hydrieranlagen in Baton Rouge (Louisiana) und eine in Baytown (Texas). Das seinerzeit größte Steinkohle-Hydrierwerk der Welt ging 1935 in Billingham im Nordosten Englands in Betrieb. Während der Eröffnungsfeier betonte der britische Premierminister Ramsay MacDonald, dass der Bau von Hydrierwerken „im Interesse der nationalen Sicherheit“ sei.[32] Die Planungen für das Werk begannen 1931 und waren offiziellen britischen Angaben zufolge auf einen künftigen Krieg ausgerichtet, um bei einer Seeblockade auf einheimische Öl-Ressourcen zurückgreifen zu können.[33] Das technische Knowhow lieferte die I.G. Farben, die im Februar 1932 ein „Engineer-Agreement“ mit der ICI vereinbarte.[34][35]
Die britische Regierung subventionierte sowohl den Bau der Anlage sowie bis Oktober 1939 die gewonnenen Treibstoffe.[36][37][38] Das Werk in Billingham hatte eine Kapazität von 150.000 t/a (Tonnen Treibstoffe pro Jahr) und wurde erst 1963 stillgelegt.[39]
Auf Grundlage eines Gesetzes vom 11. Mai 1936 erließ das Ministero delle Finanze Unternehmen, die italienische Kohle durch Hydrierung zu synthetischen Erzeugnissen verarbeiteten, für die Dauer von 10 Jahren unter anderem die Einkommensteuer, die Umsatzsteuer sowie die Aktienkapitalabgabe.[40] Im gleichen Jahr begann Italien mit dem Bau von Bergius-Pier-Hydrieranlagen in Bari und Livorno. Beide Werke gingen 1938 mit einer Jahresleistung von jeweils 180.000 Tonnen in Betrieb und arbeiteten in Kombination mit vorhandenen Erdöl-Raffinerien.[41][42] In der Folgezeit wurden die Anlagen erweitert und lieferten ab 1940 jährlich 450.000 Tonnen flüssige Ölprodukte.[43]
Betreiber der Hydrierwerke war die Azienda Nazionale Idrogenazione Combustibili (ANIC), ein Zusammenschluss von Montecatini, Azienda Italiana Petroli Albanesi (AIPA) und Agip (heute Eni). Während der schweren britischen und US-amerikanischen Tages- und Nachtbombardements vom 19. Mai bis 7. Juni 1944 auf Livorno wurden das Zentrum der Stadt sowie alle umliegenden Industriegebiete vollständig zerstört. Nach der Eroberung Norditaliens ließ die alliierte Militärregierung die Reste des Hydrierwerkes demontieren.[44] Ab 1947 existierten konkrete Pläne der ANIC zum Wiederaufbau der Anlage, die jedoch zugunsten einer Raffinerie keine Anwendung fanden.[45] Hingegen blieb das Hydrierwerk in Bari bis 1974 in Betrieb und wurde erst 1976 endgültig geschlossen.[46][47]
Spanien gründete 1942 das nationale Volksunternehmen Empresa Nacional Calvo Sotelo (heute Repsol) und schloss 1944 ein Abkommen mit Deutschland zur Errichtung eines Hydrierwerkes nach dem I.G.-Verfahren in Puertollano. 1950 unterzeichnete die spanische Regierung neue Verträge mit der BASF. Die Produktion synthetischen Benzins begann hier 1956 und endete 1966. Danach erfolgte die Umstellung auf andere chemische Produkte, die in verschiedenen Repsol-Werken bis heute hydriert werden.[48]
Am 5. April 1944 verabschiedete die US-Regierung unter Federführung des United States Bureau of Mines per Gesetz das Synthetic Liquid Fuels Program und bewilligte für die nächsten fünf Jahre 30 Millionen US-Dollar (entspräche heute einer Kaufkraft von 433.160.345 Dollar[49]). Das Ziel des Programms bestand darin, „den Bau und Betrieb von Hydrieranlagen zur Herstellung synthetischer flüssiger Brennstoffe aus Kohle, Ölschiefer, land- und forstwirtschaftlichen Erzeugnissen und anderen Stoffen für die Kriegsführung zu unterstützen sowie die Öl-Ressourcen der Nation zu erhalten und zu erhöhen“.[50][51]
Zwischen 1945 und 1951 entstanden in der Nähe von Pittsburgh und in Louisiana (Missouri) zwei spezielle Forschungsanlagen. Das Werk in Louisiana produzierte 1949 unter anderem pro Tag 200 Barrel Öl nach dem Bergius-Verfahren.[52] 1953 beendete der neue republikanische Haushaltsausschuss die Finanzierung der Forschungen. Die Anlage in Louisiana blieb jedoch unter Leitung des United States Department of the Army in Betrieb. In der Folgezeit wurde in den USA unter der Bezeichnung CtL von Kellog und in Südafrika von Sasol an verschiedenen Hydrierverfahren weitergeforscht, wobei sich hier insbesondere die Fischer-Tropsch-Synthese weiterentwickelte.[53]
Im Zuge des Ölpreisschocks nahm die US-Regierung ab 1973 die Forschung und Entwicklung synthetischer Anlagen wieder auf. 1979, nach der zweiten Ölkrise, genehmigte der US-Kongress den Energy Security Act zur Gründung der Synthetic Fuels Corporation und bewilligte rund 88 Millionen Dollar für Projekte mit synthetischen Brennstoffen. Das vollständig von der US-Regierung finanzierte Unternehmen konzentrierte sich auf die Entwicklung und den Bau kommerzieller Hydrieranlagen. Neben dem Bergius-Pier-Prozess war insbesondere das Lurgi-Ruhrchemie-Verfahren von Bedeutung. Nach der Ölschwemme von 1985 wurde die Synthetic Fuels Corporation von der Reagan-Administration aufgelöst. Bis dahin sollen für die Herstellung synthetischer Kraftstoffe in den USA, gemessen nach heutiger Kaufkraft, insgesamt 8 Milliarden Dollar öffentliche Mittel verbraucht worden sein.[54][55]
Nachkriegszeit Deutschland

Nach der Beendigung des Zweiten Weltkriegs verfügten die Alliierten zunächst die Stilllegung der Hydrieranlagen in Deutschland. Die Sowjetarmee ließ die Hydrierwerke Magdeburg, Rodleben und Pölitz demontieren und in Dserschinsk in der Nähe von Gorki wieder aufbauen.[56] Dorthin verschleppte die Sowjetunion in der Aktion Ossawakim zeitgleich zahlreiche von den bis dahin in den Hydrierwerken tätigen Ingenieuren und Wissenschaftlern.[57] Gleichfalls ließ die US-Regierung im Rahmen der Operation Paperclip deutsche Ingenieure und Chemiker, vor allem der Brabag, I.G. Farben und des Kaiser-Wilhelm-Instituts für Kohlenforschung, für das Synthetic Liquid Fuels Program überwiegend nach Louisiana (Missouri) verbringen.[58][59]
Als Anfang der 1950er Jahre das Hydrierverbot aufgehoben wurde, waren die auf Erdölbasis verfügbaren Produkte so preiswert, dass sich eine Wiederinbetriebnahme in Westdeutschland nicht rentierte. In der Deutschen Demokratischen Republik wurde die Hydrierung von Braunkohleschwelölen jedoch wieder aufgenommen und die Produkte zur Devisenbeschaffung im westlichen Ausland verkauft.[60] Die letzten Braunkohlehydrieranlagen wurden 1990 in Zeitz abgestellt.
Unter dem Eindruck der Ölkrise kündigte Helmut Schmidt in einer Regierungserklärung vom Juli 1979 an, dass die Bundesregierung die Technologie der Kohlehydrierung subventionieren wolle. Veba und Ruhrkohle AG bauten daraufhin 1981 in Bottrop eine Versuchsanlage für die Hydrierung von 200 Tonnen Kohle pro Tag, deren Betrieb 1993 eingestellt wurde.[61]
Anwendungen in der Gegenwart

Durch den niedrigen Ölpreis wurden bis Ende der 1990er Jahre in Europa, Japan, Russland und den Vereinigten Staaten keine neuen Projekte zur Kohlehydrierung verfolgt.[62] Laut damaliger Angaben der RAG Aktiengesellschaft lohne sich das Verfahren erst, wenn der Benzinpreis bei 2,30 DM liege.[63]
Infolge stark schwankender Erdölpreise gewinnen Hydrierwerke unter Anwendung verschiedener Technologien seit Beginn des 21. Jahrhunderts weltweit wieder an Bedeutung.[64] Analysten zufolge wird die Schwelle zur Wirtschaftlichkeit ab einem Ölpreis von 60 US-Dollar je Barrel überschritten.
Im Jahr 2003 baute die Shenhua Coal Liquefaction and Chemical Co. mit einem Investitionsvolumen von über zwei Milliarden Dollar ein Hydrierwerk in Ejinhoro-Banner, das 2009 den Testbetrieb aufnahm. Die dortigen Kohlereserven in der Gegend von Ordos betragen nach Schätzungen 160 Milliarden Tonnen Kohle. Für das Jahr 2013 meldete Shenhua einen Ausstoß des Hydrierwerks von 866.000 Tonnen Ölprodukten.[65]
Rohstoffe
Wasserstoff
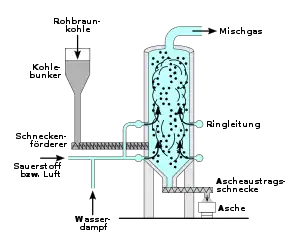
Die Erzeugung des Wasserstoffs erfolgte über die Kohlevergasung mit Hilfe eines Winkler-Generators im Wirbelbett. In einer Reihe von exothermen und endothermen Reaktionen reagiert dabei kleingemahlene Kohle mit Sauerstoff und Wasser zu Kohlenstoffdioxid und Wasserstoff. Der Einsatz von reinem Sauerstoff gegenüber Luft vermeidet bei der späteren Hydrierung die unerwünschte Senkung des Wasserstoffpartialdrucks durch Luftstickstoff. Die Sauerstoffgewinnung der damals betriebenen Luftzerlegungsanlagen diente fast ausschließlich diesem Zweck.[66] Die feine Zerteilung der Kohle gewährte einen guten Wärmeübergang, die Temperatur innerhalb des Generators war in etwa konstant. Durch die Fahrweise fiel kein Teer an, das gewonnene Gas war frei von Kohlenwasserstoffen. Da die Betriebstemperatur des Wirbelbettvergasers unter dem Schmelzpunkt der Asche gehalten werden musste, eignete sich der Winkler-Generator besonders zur Vergasung von reaktiven Kohlesorten.[67]
Die Verbrennung von Kohle mit Sauerstoff zu Kohlenstoffmonoxid (Reaktion 1) lieferte die Energie für die endotherme Erzeugung von Wassergas (Reaktion 2). Kohlenstoffmonoxid reagiert in einer reversiblen Reaktion zu Kohlenstoff und Kohlenstoffdioxid (Reaktion 3). Das sich je nach Temperatur und Druck einstellende Gleichgewicht zwischen den Reaktanden wird nach dem französischen Chemiker Octave Leopold Boudouard als Boudouard-Gleichgewicht bezeichnet. Der entstehende Kohlenstoff steht wieder der Verbrennung zur Verfügung. Kohlenstoffmonoxid kann weiterhin in einer reversiblen Reaktion, der so genannten Wassergas-Shift-Reaktion, mit Wasser zu Kohlenstoffdioxid und Wasserstoff reagieren (Reaktion 4).[68]

Eine Druckadsorptionsstufe entfernt das entstehende Kohlenstoffdioxid aus dem Gasgemisch. Nach diesem Schritt steht für die Hydrierung reiner Wasserstoff zur Verfügung.
Braunkohle

Braunkohle ist ein fossiler Brennstoff mit einem Kohlenstoffgehalt von etwa 58 bis 73 %. Sie ist in der Regel geologisch jünger als Steinkohle und weist einen geringeren Inkohlungsgrad auf. Der Wassergehalt von Rohbraunkohle beträgt je nach Herkunft 15 bis 60 %, der Gehalt an anorganischen Aschebestandteilen liegt zwischen 3 und 20 %.[69] Gegenüber Steinkohle weist Braunkohle einen höheren Sauerstoffgehalt auf, der durch Huminsäuren verursacht wird. Der hohe Gehalt an flüchtigen Bestandteilen in der Braunkohle vereinfacht die Überführung in flüssige Produkte durch Hydrierung.[69]
Braunkohlenteer
Braunkohlenteer entsteht bei der Schwelung von Braunkohle. Der Teer besteht überwiegend aus aliphatischen Kohlenwasserstoffen. Beim Einsatz von Braunkohlenteer ist eine vorherige Destillation erforderlich, die leichter flüchtige Bestandteile mit einem Siedepunkt von bis zu 325 °C abtrennt. Die Hydrierung des Destillats erfolgt in der Gasphasenhydrierung, während der Rückstand in der Sumpfphase hydriert wird.[70]
| Gehalt an Heteroelementen und Wasserstoff sowie der Wasserstoffbedarf verschiedener Rohstoffe bei der Hydrierung[71] | |||
|---|---|---|---|
| Rohstoff | Gehalt an Heteroelementen pro 100 g Kohlenstoff |
Wasserstoffgehalt pro 100 g Kohlenstoff |
Wasserstoffbedarf in Liter pro Kilogramm Benzin |
| Esskohle | 5,8 | 4,8 | 1210 |
| Gasflammkohle | 12,6 | 6,5 | 1145 |
| Schwelteer (Gasflammkohle) |
14,1 | 10,3 | 670 |
| Braunkohle | 39,2 | 7,6 | 1350 |
| Braunkohleschwelteer | 9,7 | 12,2 | 1030 |
| Erdöl | 1,9 | 14,7 | 246 |
Steinkohle
Von den Steinkohlen wurde bevorzugt Gasflammkohle eingesetzt, die durch Hydrierung bis zu 73 % flüssige Kohlenwasserstoffe lieferte.[72] Bezogen auf wasser- und aschefreie Kohle besteht Gasflammkohle zu etwa 80 % aus Kohlenstoff, 5 % aus Wasserstoff, 12 % aus Sauerstoff, 1,5 % aus Stickstoff und 1,5 % aus Schwefel.[73] Der Anteil der flüchtigen Bestandteile liegt bei 40 %. Der Wassergehalt liegt mit 10 % deutlich unter dem der Braunkohle. Die Vorbereitung der Kohle wird dadurch erleichtert und der Energiebedarf gegenüber Braunkohle ist dementsprechend geringer. Auch der Aschegehalt ist deutlich niedriger als der von Braunkohle.[73]
Steinkohlenschwelteer
Steinkohlenteer oder Tieftemperaturteer entsteht bei der Schwelung von Steinkohlen bei Temperaturen unterhalb von 700 °C. Bevorzugt wurden Kohlesorten mit einem hohen Anteil an flüchtigen Stoffen, wie Gasflammkohle, verwendet. Dabei wurde die Kohle fein zerkleinert einem Schwelofen von oben zugegeben. Als festes Produkt wurde am Boden des Schwelofens Schwelkoks ausgetragen. Die leichtflüchtigen Bestandteile wurden am Schwelofenkopf abgezogen und zum Teil als Spülgas zirkuliert. Das Spülgas wurde durch Verbrennungsgase aufgeheizt. Der größte Teil der flüchtigen Bestandteile wurde abgezogen und in Entteerungsanlagen verflüssigt und in einem Ölwäscher von anfallendem Benzin getrennt. Das Produktionsvolumen betrug 1944 etwa 200.000 Tonnen Steinkohlenschwelteer.[74]
Katalysator
Das ursprünglich von Bergius entwickelte Verfahren verwendete keine speziellen Katalysatoren, sondern nutzte die katalytischen Eigenschaften der anorganischen Bestandteile der Kohleasche. Die Asche bestand aus Oxiden, Sulfaten, Silikaten und Phosphaten verschiedener Elemente, meist Eisen und Aluminium, sowie Alkali- und Erdalkaliverbindungen.[75] Die mit dieser Methode erreichten Kohleumsätze blieben meist unbefriedigend.
Katalysatoren für die Sumpfphasenhydrierung

Die Entwicklung eines geeigneten Katalysators erwies sich aufgrund der Vielzahl der eingesetzten Rohstoffe, der Schwankungen in der Zusammensetzung des jeweiligen Rohstoffs sowie dem hohen Gehalt an Heteroelementen als schwierig. Als Katalysator für die Sumpfphasenhydrierung setzte Matthias Pier zunächst Bayermasse ein, zum Teil in Verbindung mit Goethit. Bayermasse fiel in großen Mengen als preiswertes Nebenprodukt bei der Gewinnung von Aluminiumoxid aus Bauxit im Bayer-Verfahren an (Rotschlamm). Neben Eisenoxiden enthält Bayermasse in größerer Menge Titanoxid, Aluminiumoxid und Siliziumdioxid, daneben Oxide oder Hydroxide von Natrium, Calcium, Chrom, Magnesium, Kupfer und weiteren Metallen.[76] Dieser Katalysator verhielt sich unempfindlich gegen die in der Braunkohle enthaltenen Schwefelverunreinigungen und den bei der Hydrierung entstehenden Schwefelwasserstoff.[77] Die mit Bayermasse erzeugten Kohleumsätze lagen im Bereich von etwa 50 % und damit noch zu niedrig für eine großtechnische Anwendung. Außerdem verkokten die Reaktoren stark, so dass diese mechanisch von nicht umgesetztem Koks befreit werden mussten.
Ein von Pier entwickelter Katalysator auf Basis von Molybdänoxid (MoO3), Zinkoxid und Magnesiumoxid, welcher als Pulver zur Sumpfphase gegeben wurde, steigerte den Umsatz deutlich. Der Bedarf an teurem Molybdänoxid, das zwar zum Teil zurückgewonnen wurde, erhöhte jedoch die Kosten des Verfahrens spürbar.[77] Außerdem setzte sich die spezifisch schwerere Katalysatorphase zum Teil am Reaktorboden ab und nahm damit an der katalytischen Umsetzung nur noch bedingt teil. Auf der Suche nach preiswerteren Alternativen zeigte sich mit Eisensulfat und Natronlauge imprägnierter Staub aus den Zyklonen des Winkler-Generators als hochaktiv. Der Staub bestand zu etwa 65 % aus Kohlenstoff und wurde als Ölmaische eingesetzt. Bezogen auf die eingesetzte Kohle war ein etwa 10%iger Katalysatoranteil notwendig, um einen Kohleumsatz von etwa 90 % zu erzielen. Der Effekt der feinen Verteilung des Katalysators durch den Winkler-Staub spielte eine wichtige Rolle bei der Erhöhung der katalytischen Aktivität. Die Berührung zwischen der Kohle und dem Katalysator intensivierte sich dadurch. Der Katalysator verblieb außerdem in der Kohle-Öl-Phase und setzte sich nicht ab.[78] Diese 1928 von Pier entwickelte Katalysatorvariante wurde bis zur Einstellung des Verfahrens im Jahr 1959 eingesetzt.[77]
Katalysatoren für die Gasphasenhydrierung
Als einen der ersten Katalysatoren für die Vorhydrierung zur Umsetzung der Heteroverbindungen setzte Pier den in der Sumpfphase erfolgreich getesteten Katalysator auf Basis von Molybdänoxid, Zinkoxid und Magnesiumoxid ein. Die noch vorhandenen Stickstoffverbindungen und das daraus entstehende Ammoniak deaktivierten den in Form von Würfeln eingesetzten Kontakt jedoch schnell.[77]
Erst ein Kontakt auf Basis von Wolframsulfid zeigte eine ausreichende Langzeitaktivität für die Vorhydrierung. Der aus Ammoniumparawolframat gefällte und mit Schwefelwasserstoff geschwefelte Kontakt zerfiel bei der anschließenden thermischen Zersetzung zu einem überstöchiometrischen Wolframsulfid der Form WS2,15. Jedoch erwies sich die Hydrieraktivität des Kontakts als sehr hoch. So hydrierte der Kontakt Benzol zu Cyclohexan und erniedrigte somit die Oktanzahl der Produkte.[77] Die Suche nach preiswerteren und weniger aktiven Kontakten führte zum Einsatz von Kontakten auf Basis von Nickelsulfid in Kombination mit Wolframsulfid oder Molybdänsulfid auf Aluminiumoxid.[77] Dieser Katalysatortyp entspricht den Hydrodesulfurierungskatalysatoren, die später in Erdölraffinerien eingesetzt wurden.[79]
Für die zweite Stufe der Gasphasenhydrierung, der Benzinierung, waren Kontakte erwünscht, die eine gute Hydrocrack- und Isomerisierungsaktivität aufwiesen. Aus Untersuchungen zur Isomerisierung von Paraffinen war die Eignung von säurebehandelten Alumosilicaten als Katalysator für diese Reaktion bekannt. Schließlich kam ein Wolframsulfid auf fluorwasserstoff-aktivierter Bleicherde-Kontakt als Katalysator für die Benzinierung zum Einsatz.[77]
Reaktionstechnik

Das Bergius-Pier-Verfahren lässt sich in die Teilschritte Kohlebreiherstellung, Sumpfphasenhydrierung und Gasphasenhydrierung unterteilen. Die Gasphasenhydrierung bestand aus der Vorhydrierung und der Benzinierung. Die Aufarbeitung der Produkte erfolgte destillativ. Zur Entfernung von Schwefelwasserstoff und Kohlenstoffdioxid wurde die Gasphase einer Alkazid-Wäsche unterworfen. Dabei bildete Kalium-N,N-dimethylglycinat, das Kaliumsalz des Dimethylglycins, bei Raumtemperatur ein Addukt mit Schwefelwasserstoff oder Kohlenstoffdioxid, welches bei Temperaturen von oberhalb 100 °C wieder in die Ausgangsprodukte zerfiel.[80]
Kohlebreiherstellung

Zunächst wurde die Braunkohle in einer Hammermühle auf eine Korngröße von fünf Millimetern gemahlen und danach auf einen Wassergehalt von 4 % getrocknet. Vor dem Trocknen wurde Bayermasse zur Braunkohle gegeben und ein Eisengehalt von etwa 2,5 % eingestellt. Nach der Trocknung erfolgte eine Nachmahlung auf eine Korngröße von einem Millimeter.[70]
Nach Zugabe von etwa 15 % Anreibeöl wurde die Braunkohle in einer Breimühle zu Kohlebrei verarbeitet. Das Arbeiten unter Stickstoff als Schutzgas minimierte die Oxidation der Braunkohle. Mittels einer Breipresse wurde die Masse im nächsten Schritt zu einem Wärmetauscher, dem Regenerator, gepumpt, wobei mit weiterem Anreibeöl ein Feststoffgehalt von 48 % eingestellt wurde. Der Kohlebrei enthielt typischerweise einen Aschegehalt von etwa 20 %.[70]
Sumpfphasenhydrierung
Der Kohlebrei, wegen des hohen Feststoffgehalts als Sumpf bezeichnet, wurde bei Temperaturen von 450 bis 500 °C und Wasserstoffdrücken von 200 bis 700 bar in einer exothermen Reaktion hydriert. Dabei erfolgte die fast vollständige Konvertierung der Heteroatome der in der Kohle enthaltenen Organoschwefel-, Organostickstoff- und Organosauerstoffverbindungen in ihre flüchtigen Wasserstoffverbindungen. Weiterhin fand hier die Spaltung und Sättigung der Kohlenwasserstoffe statt.[70]
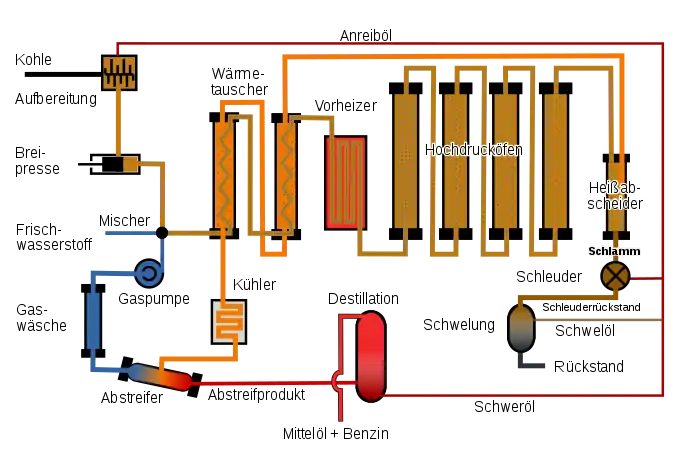
Zunächst förderte eine Breipresse den Kohlebrei über zwei Wärmetauscher, Regeneratoren genannt, und einen Vorheizer zum Hochdruckofen. In den Regeneratoren fand die Vorwärmung des Kohlebreis mit heißen Produkten aus der Hydrierung statt. Im Vorheizer erfolgte die Beheizung mit Gas bis auf die Reaktionstemperatur von 450 bis 500 °C.[70]
Die Verarbeitung von einem Kubikmeter Kohlebrei pro Stunde erforderte in etwa dasselbe Reaktorvolumen. Ein typischer Reaktor, der Hochdruckofen, hatte einen Durchmesser von etwa einem Meter bei einer Höhe von 12 bis 18 Metern und ein Volumen von etwa neun Kubikmetern. Bei einer Verarbeitungskapazität von 250 Kubikmetern Kohlebrei pro Stunde verbrauchte die Hydrierung etwa 86.000 Normkubikmeter Wasserstoff. Dazu wurden 360.000 Normkubikmeter Wasserstoff zirkuliert, zum Teil um den Kohlebrei zu durchmischen, zum Teil unter Zugabe von kaltem Wasserstoff, der die Hydrierwärme aufnahm.[70] Im zirkulierten Wasserstoff reicherten sich Methan und Ethan sowie Stickstoff, Kohlenstoffmonoxid und Kohlenstoffdioxid an. Um eine Senkung des Wasserstoffpartialdrucks durch diese Gase zu minimieren, wurde das zirkulierte Gas vor dem Eintritt in den Reaktor einer Ölwäsche bei 250 bar unterzogen. Dabei lösten sich die Kohlenwasserstoffe im Mittelöl, das aus der Hydrierung stammte.[70]
Das Mittelöl wurde in zwei Stufen entspannt. In der ersten Druckentspannungsstufe von 25 bar wurden die niedermolekularen Gase wie Wasserstoff, Methan, Kohlenstoffmonoxid und Kohlenstoffdioxid freigesetzt. In der zweiten Entspannungsstufe bei Normaldruck wurden die Flüssiggase sowie etwas Pentan freigesetzt.[70]
Die Produkte der Sumpfphasenhydrierung ähnelten in ihrer chemischen Struktur noch den eingesetzten Kohlen. Steinkohlenöle enthielten viele aromatische Verbindungen, aus Braunkohle gewonnene Öle enthielten überwiegend aliphatische Kohlenwasserstoffe. Die Trennung der entstandenen Öle von unhydrierbaren Bestandteilen der Kohle erfolgte durch Destillation.[70] Der Katalysator verblieb in den nicht hydrierbaren Bestandteilen. Diese nicht destillierbaren Feststoffe, die reich an Asche sowie Katalysatorbestandteilen waren, konnten in der Kohlevergasung wieder zur Herstellung von Wasserstoff genutzt werden.
Gasphasenhydrierung
Die Gasphasenhydrierung unterteilt sich in eine Vorhydrierung und die so genannte Benzinierung. In der Stufe der Vorhydrierung erfolgt die Entfernung der noch nicht in der Sumpfphase eliminierten Heteroelemente. Die Vorhydrierung wurde notwendig, da die in der Benzinierung verwendeten sauren Katalysatoren sehr empfindlich gegen eine Vergiftung durch Ammoniak oder andere basische Stickstoffverbindungen waren.[71] In der Vorhydrierung wurde das so genannte A-Mittelöl, das einen hohen Stickstoffgehalt aufwies, ohne Spaltung hydroraffiniert. Nach der Abtrennung der Benzinfraktion entstand das so genannte, fast stickstofffreie B-Mittelöl, das in der Benzinierung einer Hydrocrackreaktion unterworfen wurde.[77]
In der Benzinierungsstufe entstanden die Zielprodukte, heteroelementfreie Kohlenwasserstoffe mit dem geforderten Siedebereich sowie der richtigen Viskosität und Oktanzahl. Die Gasphasenhydrierung verbrauchte etwa 25 % des Gesamtwasserstoffs. Isomerisierungsreaktionen liefen ohne Wasserstoffbedarf ab und die Dehydrierung von Naphthenen zu Aromaten unter Freisetzung von Wasserstoff.[71]
Betriebsdaten des Hydrierwerks Wesseling
Das Werk Wesseling hatte ein nominale Kapazität von 260.000 Tonnen pro Jahr. Den höchsten Ausstoß erreichte das Werk 1943, als der Ausstoß 39.400 Tonnen Autobenzin, 93.200 Tonnen Flugbenzin, 72.800 Tonnen Dieselkraftstoff und 21.100 Tonnen Treibgase betrug. Daneben erzeugte das Werk 1000 Tonnen Phenol.[71]
Als Rohstoff diente Braunkohle. Die Wasserstofferzeugung erfolgte zum Teil aus Braunkohle, wobei pro Stunde 47.500 Normkubikmeter erzeugt wurden, und zum Teil aus Methan und anderen leichten Kohlenwasserstoffen, mit denen pro Stunde 36.500 Normkubikmeter Wasserstoff gewonnen wurden. Die Konvertierung erfolgte mittels Eisenoxid/Chromoxid-Kontakten mit anschließender Kohlenstoffmonoxid- und Kohlenstoffdioxid-Wäsche.[71]
Die Sumpfphasenhydrierung erfolgte in vier Reaktorkammern mit einem Gesamtvolumen von 32 Kubikmetern, die bei einer Temperatur von 475 °C und 650 bar betrieben wurden, wobei der Wasserstoffpartialdruck 475 bar betrug. Die Braunkohle wurde als Brei aus 36 % Braunkohle mit 6,25 % Katalysator in Anreibeöl zu den Reaktoren gepresst. Das dort gebildete Mittelöl bestand nach der Entfernung der Phenole zu etwa zwei Dritteln aus Aromaten, der Rest verteilte sich auf Olefine, Naphthene und Paraffine.[71]
Die Vorhydrierung wurde in acht Reaktoren mit einem Gesamtvolumen von 64 Kubikmetern durchgeführt. Drei Reaktoren waren dabei mit Wolframsulfid-Kontakten bestückt und fünf Reaktoren mit einem Nickelsulfid/Molybdänsulfid/Wolframsulfid auf Aluminiumoxid-Kontakten.[71] Die Benzinierung erfolgte in fünf Reaktoren mit einem Gesamtvolumen von 40 Kubikmetern, in denen fluorwasserstoff-aktiviertes Wolframsulfid auf Bleicherde-Kontakt eingesetzt wurde. Die Vorhydrierung- und die Benzinierungsstufe benötigten insgesamt 620 Normkubikmeter Wasserstoff pro Tonne Rohstoffdurchsatz.[71]
Produkte
Das Produktspektrum hing von der chemischen Zusammensetzung der eingesetzten Kohle ab, etwa dem Inkohlungsgrad oder dem Aschegehalt, sowie von den Reaktionsbedingungen wie dem Wasserstoffdruck, der Temperatur und der Verweilzeit. Braunkohle liefert überwiegend paraffinische Produkte, die als Dieselkraftstoff verwendet werden, während Steinkohle höheroktanige, aromatische Produkte für die Verwendung als Motorenbenzin liefert.[81]
Gase
Als gasförmige Erzeugnisse fielen Methan, Ethan, Propan und ein Gemisch von Butan und Isobutan an. Die Produktion von 100.000 Tonnen Benzin aus Braunkohle erzeugte etwa 23.000 Tonnen Flüssiggase, davon 10.000 Tonnen Propan und 13.000 Tonnen eines Butan/Isobutan-Gemischs aus. Im Winter verblieben etwa 5000 Tonnen des Butans im Benzin, 8000 Tonnen standen der chemischen Industrie zur Verfügung. Daneben fielen noch etwa 6500 Tonnen Ethan an.[70] Die gasförmigen Kohlenwasserstoffe bildeten sich durch Crackreaktionen, wobei zunächst Alkene entstanden, die sofort weiter zu den entsprechenden Alkanen hydriert werden.
Die entstandenen gasförmigen Kohlenwasserstoffe wurden in Arm- und Reichgase unterschieden. Die Armgase enthielten neben Wasserstoff vor allem Methan und einen Teil des Ethans, die Reichgase enthielten vor allem Flüssiggase und kaum Wasserstoff. Die Gase wurden vor allem durch die Entspannung von Waschölen, bei der Destillation der Produkte und bei der Benzinierung erhalten.[70]
Benzin
Das Bergius-Pier-Verfahren lieferte unabhängig vom Rohstoff und den Verfahrensparametern ein paraffin- und naphthenreiches Benzin mit einer Motoroktanzahl von 71 bis 73. Der Olefingehalt lag unter 1 %, der Aromatengehalt lag je nach Rohstoff zwischen 8 und 9 %.[71] Bei gleichen Rohstoffen und Verfahrensparametern erzeugten Wolframsulfid/Nickelsulfid auf Tonerde-Kontakte in der Vorhydrierung und Wolframsulfid auf Tonerde-Kontakte in der Benzinierungsstufe die Benzine mit den höchsten Oktanzahlen. Eine Aromatisierungsstufe mit Chromoxid/Vanadiumpentoxid auf Aktivkohle-Kontakte steigerte die Oktanzahl auf etwa 83.[71]
| Typische Analysenwerte von Flugbenzinen verschiedener Hydrierwerke[71] | ||||
|---|---|---|---|---|
| Parameter | Leuna (Braunkohle) |
Scholven (Steinkohle) |
Gelsenberg (Steinkohle) |
Pölitz (Steinkohle) |
| Dichte in g/cm³ | 0,719 | 0,738 | 0,740 | 0,730 |
| Siedebeginn in °C | 45 | 44 | 46 | 44 |
| Siedeende in °C | 139 | 156 | 151 | 152 |
| % Paraffine | 51,5 | 37,5 | 36,5 | 48,5 |
| % Naphthene | 38 | 53 | 54 | 43 |
| % Aromaten | 8,5 | 8,5 | 9 | 7,5 |
| % Olefine | 1 | 1 | 0,5 | 1 |
| MOZ | 71 | 73 | 73 | 72 |
Dieselkraftstoff
Die erzeugten Dieselkraftstoffe aus Braunkohle wiesen gegenüber denen aus Steinkohle eine erhöhte Cetanzahl auf, Dieselkraftstoffe aus Anlagen mit einem höheren Verfahrensdruck waren paraffinreicher und damit zündwilliger.[71]
Die Dichte des Diesels lag zwischen 0,8 und 0,88 g/cm³. Die Cetanzahl betrug zwischen 45 und 55 für die Diesel, die bei Drücken von 200 bis 300 bar hydriert wurden, und zwischen 72 und 75 für Diesel, die bei hohen Drücken von 600 bar hergestellt wurden. Der Wasserstoffgehalt lag bei etwa 14 %.[71]
Mechanismus
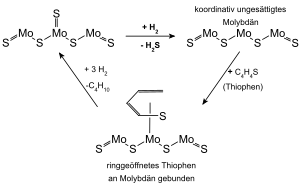
Studien von Modellsubstanzen wie Thiophen, Phenol oder Pyridin in Hydrofiningreaktionen legten nahe, dass die katalytisch aktiven Stellen der Kontakte auf den Ecken und Kanten der Katalysatorkristallite liegen.[82] Die Reaktion von Wasserstoff mit oberflächengebundenem Sulfidschwefel unter Freisetzung von Schwefelwasserstoff schafft eine koordinativ ungesättigte Oberflächenstelle, an der sich heteroatomhaltige Substrate binden können. Unter Bildung eines neuen Sulfidschwefel und eines ungesättigten organischen Restes beginnt der katalytische Zyklus von Neuem.
Beim Bergius-Pier-Verfahren laufen parallel Hydrierungs-, Hydrocrack- und Hydrorefiningreaktionen wie die Hydrodenitrogenierung und die Hydrodesulfurierung ab. Die Hydrofiningreaktionen laufen nach den folgenden Reaktionsschema ab:

Die Hydrierung überführt ungesättigte in wasserstoffreichere Kohlenwasserstoffe, Hydrocrackreaktionen reduzieren die molare Masse und führen zu leichtflüssigeren Produkten. Die Hydrofiningreaktionen eliminieren die Heteroatome Sauerstoff, Stickstoff und Schwefel im Produkt und generieren Wasser, Ammoniak und Schwefelwasserstoff. Crackreaktionen generieren niedermolekulare Alkene, die aufgrund des hohen Wasserstoffdrucks sofort zu Alkanen hydriert werden. Weiterhin setzen sie Kohlenstoffmonoxid und Kohlenstoffdioxid frei, indem sie Ester- oder andere sauerstoffhaltige funktionelle Gruppen des Ligningerüsts der Braunkohle abbauen. Die Gesamtheit der ablaufenden Prozesse wurde zeitweilig als Berginisieren der Kohle bezeichnet.[83]
Beispiele verschiedener Hydrierungs- und Hydrotreating-Reaktionen zeigt das folgende Reaktionsschema an einem Braunkohlefragment:
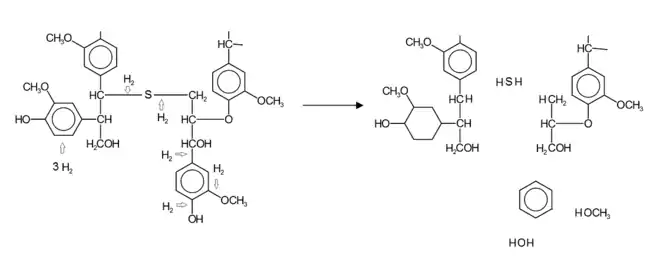
Verfahrensvarianten
Das ursprüngliche Bergius-Verfahren arbeitete ohne speziell zugesetzte Katalysatoren, sondern nutzte die in der Kohleasche vorkommenden Eisenverbindungen als Katalysatoren. Von Bergius stammte die Idee, die Braunkohle mit Öl zu einem Kohlebrei zu verreiben und diese unter hohem Wasserstoffdruck und Temperaturen von etwa 500 °C zu hydrieren. Die Verfahrensvarianten unterscheiden sich in der Reaktionsführung, dem Katalysator und der Wasserstoffquelle.
Pott-Broche-Prozess
Anfang der 1930er Jahre entwickelten Alfred Pott und Hans Broche ein Verfahren, bei dem mit Tetralin und Decalin als wasserstoffabgebenden Lösungsmittel gearbeitet wird. Tetralin und Decalin werden dabei zum Naphthalin oxidiert, welches destillativ abgetrennt und nach Hydrierung wiederverwendet werden kann. Als weiteres Lösungsmittel wird Kresol oder Phenol verwendet.[84]
Die Hydrierung wurde bei Temperaturen zwischen 415 und 435 °C und einem Druck von etwa 100 bar durchgeführt. Eine Anlage der Ruhröl produzierte zwischen 1938 und 1944 30.000 Tonnen Kohleöl, welches als Ersatz für schweres Heizöl in Kraftwerken Verwendung fand.
H-Coal-Verfahren
Beim 1963 entwickelten H-Coal-Verfahren der Hydrocarbon Research Inc. (HRI) wird Braunkohle in einem einstufigen Verfahren mit Hilfe eines Cobalt-Molybdän-Katalysators hydriert. Der Katalysator wird zur Vermeidung der Deaktivierung in einem „ebullated bed“ (wallendes Bett = Wirbelschichtreaktor) ständig in Bewegung gehalten, zum Teil ausgeschleust und durch frischen Katalysator ersetzt.[4] Die verschiedenen Crack- und Hydrierungsreaktionen laufen bei kurzer Reaktionszeit in nur einem Reaktor ab, die Produkte weisen ein hohes Wasserstoff-zu-Kohlenstoff-Verhältnis auf.
Mit finanzieller Unterstützung in Höhe von 300 Millionen USD durch das Energieministerium der Vereinigten Staaten, den US-Bundesstaat Kentucky und verschiedener Ölfirmen baute HRI eine Pilotanlage für einen Kohledurchsatz von 200 bis 600 Tonnen pro Tag.[85]
Synthoil-Verfahren
Das Synthoil-Verfahren wurde ab 1969 im Auftrag der Energy Research and Development Administration/Fossil Energy (ERDA/FE), heute Teil des Energieministeriums der Vereinigten Staaten, entwickelt. Die Anmaischung der Kohle erfolgte mit einem Lösungsmittel. Als Katalysator fand ein Cobalt-Molybdän-Katalysator Verwendung, der bei einer Temperatur von 425 bis 450 °C und einem Druck von bis zu 280 bar arbeitete. Zielprodukte waren flüssige Brennstoffe für den Einsatz in Kraftwerken.[4] Durch nicht gelöste Probleme mit der Langzeitaktivität des Katalysators wurde das Verfahren bislang nur im Technikumsmaßstab getestet.
Shenhua Direct Coal Liquefaction Process
Beim Shenhua Direct Coal Liquefaction Process wird bituminöse Kohle mit einem hohen Gehalt an inerten Bestandteilen hydriert. Die in der Inneren Mongolei gebaute Anlage ist nach dem Zweiten Weltkrieg die einzige kommerziell betriebene Kohlehydrieranlage der Welt.[86] Das Verfahren besteht im Wesentlichen aus zwei rückvermischten Reaktorstufen sowie einem Festbett-Hydrotreater. Als Katalysator wird ein feingemahlener Eisenkatalysator verwendet. Das Verfahren arbeitet bei einem Druck von 170 bar und einer Temperatur von etwa 450 °C, wobei ein Umsatz auf die eingesetzte Kohle von über 90 % erreicht wird. Die erhaltenen Produkte wie Naphtha, Dieselöl und Flüssiggas sind weitgehend frei von Schwefel und Stickstoff.[87]
Literatur
- Walter Krönig: Die katalytische Druckhydrierung von Kohlen, Teeren und Mineralölen (Das I.G.-Verfahren von Matthias Pier). Springer Verlag, 1950. (Reprint: 2013, ISBN 978-3-642-50105-0)
Weblinks
Einzelnachweise
- Heinz-Gerhard Franck: Kohleveredlung: Chemie und Technologie. Verlag Springer, 1979, ISBN 3-540-09627-2, S. 228.
- Anthony N. Stranges: Friedrich Bergius and the Rise of the German Synthetic Fuel Industry. In: Isis. 75, 1984, S. 643–667, doi:10.1086/353647.
- Emil Fischer: Die Aufgaben des Kaiser-Wilhelm-Instituts für Kohlenforschung. In: Stahl und Eisen. 32, 1912, S. 1898–1903.
- K. F. Schlupp, H. Wien: Herstellung von Öl durch Hydrierung von Steinkohle. In: Angewandte Chemie. 88, 1976, S. 347–353, doi:10.1002/ange.19760881103.
- Robert Haul: Das Portrait: Friedrich Bergius (1884–1949). In: Chemie in unserer Zeit. 19, 1985, S. 59–67, doi:10.1002/ciuz.19850190205.
- Paul Walden: Geschichte der organischen Chemie seit 1880. Springer Verlag, 1972, ISBN 3-662-27210-5, S. 847.
- Friedrich Bergius – Nobel Lecture: Chemical Reactions under High Pressure. Nobelprize.org. Nobel Media AB 2014. Web. 2 Sep 2016 (online).
- Federal Oil Conservation Board: Public Hearing – May 27, 1926. To afford full opportunity to representatives of the oil industry to present final views relating to national petroleum conditions before completion by the Government oil board of its preliminary report on this subject to the President. Government Printing Office, Washington, DC 1926 (handle.net [abgerufen am 18. Januar 2021]).
- Franz Spausta: Treibstoffe für Verbrennungsmotoren. Springer Verlag, 1939, S. 54. (Reprint: ISBN 978-3-7091-5161-7)
- Karl Becker: Die Geburt der Hochdruckchemie und das Leuna Werk: Ein Stück Geschichte der technischen Chemie – Teil 1. In: Chemie Ingenieur Technik. 85, 2013, S. 1824–1834, doi:10.1002/cite.201300096.
- Rainer Karlsch, Raymond G. Stokes: Faktor Öl: die Mineralölwirtschaft in Deutschland 1859–1974. Verlag C.H. Beck, 2003, ISBN 3-406-50276-8, S. 136.
- Matthias Pier: Der Weg zum deutschen Benzin. Niederschrift eines Rundfunkvortrags anlässlich seines 60. Geburtstags am 22. Juli 1942 (online) (Memento vom 25. September 2016 im Internet Archive) (PDF)
- Werner Abelshauser: Die BASF, eine Unternehmensgeschichte. Verlag C.H. Beck, 2003, ISBN 3-406-49526-5, S. 206.
- Emanuel Heisenberg: Momente der Entscheidung: Tödliche Synthese. In: Die Zeit. 3. Juni 2004 (online).
- Werner Abelshauser: Die BASF, eine Unternehmensgeschichte. Verlag C.H. Beck, 2003, ISBN 3-406-49526-5, S. 238–242.
- Judith Mirzoeff: Cracking the oil from coal Problem. In: New Scientist. 17. Juli 1980, S. 223.
- Titus Kockel: Deutsche Ölpolitik 1928–1938. Akademie-Verlag, 2005, ISBN 3-05-004071-8, S. 100.
- Wilhelm Treue: Hitlers Denkschrift zum Vierjahresplan 1936. In: VfZ. 2/1955, S. 184 ff. (online (PDF; 1,3 MB). Abgerufen am 10. September 2016)
- Günter Bayerl: Braunkohleveredelung im Niederlausitzer Revier. 50 Jahre Schwarze Pumpe. Waxmann Verlag, 2009, ISBN 978-3-8309-1684-0, S. 63.
- Karl Heinz Blumenhagen: Die deutsch-sowjetischen Handelsbeziehungen 1939-1941und ihre Bedeutung für die jeweilige Kriegswirtschaft. Verlag Dr. Kovač, 1998, S. 212.
- Heinz-Gerhard Franck, Jürgen Walter Stadelhofer: Industrielle Aromatenchemie: Rohstoffe, Verfahren, Produkte. Verlag Springer, 1987, ISBN 3-662-07876-7, S. 48.
- Dieter Osteroth: Von der Kohle zur Biomasse. Springer Verlag, 1989, ISBN 3-540-50712-4, S. 18.
- Günther Luxbacher: Kohle – Öl – Benzin. In: Helmut Maier: Gemeinschaftsforschung, Bevollmächtigte und der Wissenstransfer. Wallstein-Verlag, 2007, ISBN 978-3-8353-0182-5, S. 443.
- Schlacht um Sprit. In: Der Spiegel. Nr. 6, 1964, S. 60–62 (online).
- Ralf Schabel: Die Illusion der Wunderwaffen. R. Oldenbourg Verlag, 1993, ISBN 3-486-55965-6, S. 236.
- Weltmacht Öl Der Spiegel vom 24. Dezember 1973, abgerufen am 1. Juli 2019
- Alan J. Levine: The strategic bombing of Germany 1940–1945. Verlag Praeger, 1992, ISBN 0-275-94319-4, S. 151.
- Weltmacht Öl Der Spiegel vom 24. Dezember 1973, abgerufen am 1. Juli 2019
- Christoph Gunkel: Terror unter Tage. In: Spiegel online. 28. September 2009 (online).
- Moneir Nasr: Mineralölwirtschaft im Nahen Osten. Verlag für Literatur und Zeitgeschehen, 1967, S. 29.
- Walter Wetzel: Geschichte der deutschen Chemie in der ersten Hälfte des 20. Jahrhunderts. Band 19. Mitteilungen, Fachgruppe Geschichte der Chemie, Frankfurt a.M., 2007, S. 189. Gesellschaft Deutscher Chemiker, abgerufen am 26. Juni 2019
- Michael Stratton, Barrie Trinder: Twentieth Century Industrial Archaeology. Taylor & Francis, 2014, S. 84.
- Billingham Wharf and Official Opening of the Coal Hydrogenation Petrol Plant, ICI Billingham, by James Ramsay MacDonald M.P. British Film Institute, abgerufen am 29. Juni 2019
- Walter Wetzel: Geschichte der deutschen Chemie in der ersten Hälfte des 20. Jahrhunderts. Band 19. Mitteilungen, Fachgruppe Geschichte der Chemie, Frankfurt a.M., 2007, S. 198. Gesellschaft Deutscher Chemiker, abgerufen am 26. Juni 2019
- Bruno Riediger: Die Verarbeitung des Erdöles. Springer-Verlag, 2013, S. 806.
- Franz Spausta: Treibstoffe für Verbrennungsmotoren. Springer-Verlag, 2013, S. 75.
- J. L. Wiley, H. C. Anderson: Bibliography of Pressure Hydrogenation. Bände 1-3. U.S. Government Printing Office, 1950, S. 216, 277.
- E. Beesley, B. Wipp: "Butane dehydrogenation at Billingham". Chemical Industry Press London, 1953, S. 550–556.
- E. Bartholomé: Ullmanns Encyklopädie der technischen Chemie. Band 14. Wiley Press, 1985, S. 476.
- Franz Spausta: Treibstoffe für Verbrennungsmotoren. Springer-Verlag, 2013, S. 77.
- Walter Teltschik: Geschichte der deutschen Großchemie. Entwicklung und Einfluss in Staat und Gesellschaft. John Wiley & Sons Canada Limited, 1992, S. 117.
- Walter Krönig: Die katalytische Druckhydrierung von Kohlen Teeren und Mineralölen. Das I.G.-Verfahren von Matthias Pier. Springer-Verlag, 2013, S. 261.
- B. Brockamp, P. Ramdohr (Hrsg.): Zeitschrift für praktische Geologie. Band 47. Wilhelm Knapp Verlag, 1940, S. 150.
- Beppe Leonardini, Giovanni Corozzi, Giovanni Pentagna: Apocalisse 1943/45 distruzione di una città. Nuova Fortezza Livorno 1984, S. 3 f.
- Tamara Brunnschweiler-Peterson: Die Erdölproduktion Europas. Kapri Verlag, 1949, S. 67.
- Carlo Perego: Giacomo Fauser, Guido Donegani e le origini dell’ANIC a Novara. Istituto Donegani di Novara, 2010, S. 12. Istituto Donegani di Novara, abgerufen am 30. Juni 2019
- Bari Treccani La Cultura Italiana, abgerufen am 30. Juni 2019
- British Convert Coal in WWII (Memento des Originals vom 29. Juni 2019 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. West Virginia Coal Association, abgerufen am 29. Juni 2019
- Inflationsrechner Dollar 1944 zu Dollar 2019 In: dollartimes.com, abgerufen am 1. Juli 2019
- Early Days of Coal Research U.S. Department of Energy, abgerufen am 30. Juni 2019
- Gene Whitney, Carl E. Behrens: Energy. Natural Gas. The Capitol Net Inc, 2010, S. 309.
- Richard H. K. Vietor: Energy Policy in America since 1945. Cambridge University Press, 1984, ISBN 0-521-26658-0, S. 46–52.
- Early Days of Coal Research U.S. Department of Energy, abgerufen am 30. Juni 2019
- Oil Shale. History, Incentives, and Policy. CRS Report for Congress, abgerufen am 1. Juli 2019
- A Magic Way to Make Billions Time, abgerufen am 1. Juli 2019
- Öl im Dienste der Sowjetwirtschaft. In: Die Zeit. 16. März 1950, abgerufen am 20. September 2016.
- John E. Lesch: The German Chemical Industry in the Twentieth Century. Kluwer Academic Publishers, ISBN 0-7923-6487-2, S. 390.
- Don Bongaards: A Sense of Urgency. Xlibris Corporation, 2009, S. 42.
- Anthony N. Stranges: The US Bureau of Mines's synthetic fuel programme, 1920–1950s: German connections and American advances. In: Annals of Science. 54, 2006, S. 29, doi:10.1080/00033799700200111.
- Dieter Osteroth: Von der Kohle zur Biomasse. Springer Verlag, 1989, ISBN 3-540-50712-4, S. 133.
- Dreifache Menge. In: Der Spiegel. Nr. 6, 1980, S. 45–49 (online).
- Hengfu Shui, Zhenyi Cai, Chunbao (Charles) Xu: Recent Advances in Direct Coal Liquefaction. In: Energies. 3, 2010, S. 155–170, doi:10.3390/en3020155.
- Billiges Benzin aus Bottrop Welt am Sonntag vom 1. Juni 2008, abgerufen am 17. Juni 2019
- Lexikon der Chemie: Kohlehydrierung Spektrum Wissen, abgerufen am 17. Juni 2019
- China’s first direct coal liquefaction line produces 866,000 tonnes (Memento vom 21. August 2014 im Internet Archive). Abgerufen am 17. September 2016.
- Rudolf Plank (Hrsg.), Helmuth Hausen: Handbuch der Kältetechnik: Erzeugung sehr tiefer Temperaturen. Springer Verlag, 1957, OCLC 311803448, S. 381.
- Karl-Heinz Schmidt, Ingo Romey, Fritz Mensch: Kohle, Erdöl, Erdgas: Chemie und Technik. Vogel Verlag, 1981, ISBN 3-8023-0684-8, S. 63.
- P. Powell, P. Timms: The Chemistry of the Non-Metals. Springer Science and Business Media, 1974, ISBN 0-412-12200-6, S. 86–88.
- Karl-Heinz Schmidt, Ingo Romey, Fritz Mensch: Kohle, Erdöl, Erdgas: Chemie und Technik. Vogel Verlag, 1981, ISBN 3-8023-0684-8, S. 37–38.
- Friedrich Asinger: Chemie und Technologie der Paraffin-Kohlenwasserstoffe. Akademie Verlag, 1956, DNB 450152154, S. 29–41.
- Helmut Pichler: Herstellung flüssiger Kraftstoffe aus Kohle. Report Carl Engler und Hans Bunte-Institut für Mineralöl- und Kohleforschung und Institut für Gastechnik, Feuerungstechnik und Wasserchemie der Universität Karlsruhe, 1970, S. 5–57 (online) (Memento vom 16. September 2016 im Internet Archive).
- C. Doelter: Handbuch der Mineralchemie. Halloidsalze, Fluoride, Edelgase, organische Verbindungen (Kohlen, Asphalt, Erdöl), Nachträge, Generalregister. Springer Verlag, 1931, S. 524. (Reprint: ISBN 978-3-642-49593-9)
- Walter Krönig: Die katalytische Druckhydrierung von Kohlen, Teeren und Mineralölen (Das I.G.-Verfahren von Matthias Pier). Springer Verlag, 1950, S. 56. (Reprint: ISBN 978-3-642-50105-0)
- Heinz-Gerhard Franck, Gerd Collin: Steinkohlenteer: Chemie, Technologie und Verwendung. Springer Verlag, 1968, ISBN 3-642-88259-5, S. 1.
- Karl-Heinz Schmidt, Ingo Romey, Fritz Mensch: Kohle, Erdöl, Erdgas: Chemie und Technik. Vogel Verlag, 1981, ISBN 3-8023-0684-8, S. 216.
- H. Schmitt: Comparison of Bayermasse, Luxmasse, and Lautamasse (impure iron oxides) as catalyst in coal hydrogenation. Technischer Report, Hydrierwerk Pölitz, 17. Dezember 1938, OSTI Identifier: 6122127, Report Number: TOM-75-656-658 (online).
- Karl Becker: Katalysatoren des Leuna Werkes: ein Beitrag zur Geschichte der technischen heterogenen Katalyse – Teil I: Die Entwicklungen von 1921 bis 1945. In: Chemie Ingenieur Technik. 86, 2014, S. 2105–2114, doi:10.1002/cite.201400051.
- Diwakar Garg, Edwin N. Givens: Effect of catalyst distribution in coal liquefaction. In: Fuel Processing Technology. 7, 1983, S. 59–67, doi:10.1016/0378-3820(83)90025-5.
- Ernst Münzing, Hermann Blume, Emanuel Pindur: Arbeiten zur Verbesserung der Katalysatoren für die hydrierende Raffination von Teeren und Mittelölen. In: Zeitschrift für Chemie. 2, 1962, S. 76–83, doi:10.1002/zfch.19620020304.
- I.G. Alkazid Process for Gas Purification (Memento vom 6. Januar 2015 im Internet Archive). Abgerufen am 21. September 2016.
- W. Wilke: Der heutige Stand der Hydrierung und Synthese von Kraftstoffen und ihre Bedeutung für die Weltölwirtschaft. In: Schweizerische Bauzeitung. 23, 1948, S. 587–591, doi:10.5169/seals-56815.
- Charles N. Satterfield, Michael Modell, John A. Wilkens: Simultaneous Catalytic Hydrodenitrogenation of Pyridine and Hydrodesulfurization of Thiophene. In: Industrial & Engineering Chemistry Process Design and Development. 19, 1980, S. 154–160, doi:10.1021/i260073a027.
- A. Spilker, K. Zerbe: Beitrag zur Kenntnis der chemischen Vorgänge bei der Hydrierung der Kohle nach Bergius. In: Zeitschrift für Angewandte Chemie. 39, 1926, S. 1138–1143, doi:10.1002/ange.19260393903.
- Patent US2123380: Method of producing extracts from solid carbonaceous materials. Veröffentlicht am 12. Juli 1938, Erfinder: Alfred Pott, Hans Broche.
- Sunggyu Lee: Alternative Fuels. Verlag Taylor & Francis, 1996, ISBN 1-56032-361-2, S. 175–177.
- Long Xu, Mingchen Tang, Lin’e Duan, Baolin Liu, Xiaoxun Ma, Yulong Zhang, Morris D. Argyle, Maohong Fan: Pyrolysis characteristics and kinetics of residue from China Shenhua industrial direct coal liquefaction plant. In: Thermochimica Acta. 589, 2014, S. 1–10, doi:10.1016/j.tca.2014.05.005.
- Alfred G. Comolli, Theo L.K. Lee, Gabriel A. Popper, Peizheng Zhou: The Shenhua coal direct liquefaction plant. In: Fuel Processing Technology. 59, 1999, S. 207–215, doi:10.1016/S0378-3820(99)00016-8.