Krebsimmuntherapie
Krebsimmuntherapie ist die Bezeichnung für verschiedene Methoden der Immuntherapie zur Behandlung von Krebserkrankungen.
Die klassischen Behandlungsmethoden bei Krebs sind die operative Tumorentfernung (Resektion), die Chemotherapie und die Strahlentherapie. Häufig werden zwei oder gar alle drei Therapieformen gleichzeitig bei einem Patienten angewendet. Die beiden letztgenannten Methoden haben erhebliche zytotoxische Nebenwirkungen. Die therapeutische Breite ist bei der Chemotherapie sehr gering, so dass eine hohe Dosierung – die für eine Verstärkung der Wirkung förderlich wäre – meist ausgeschlossen ist. Werden bei der Therapie aber nicht alle Zellen des Tumors und seiner Metastasen vernichtet (eradiziert), so ist die weitere Behandlung durch Resistenzbildung deutlich erschwert. Seit Jahren wird daher an neuen Therapieverfahren geforscht, die eine möglichst hohe selektive Wirkung gegen Krebszellen aufweisen. Die verschiedenen Ansätze der Krebsimmuntherapie besitzen hier ein vielversprechendes Potenzial, das – beispielsweise bei der Antikörpertherapie – auch Einzug in die klinische Praxis gehalten hat.[1]
In der Onkologie unterscheidet man bei den unterschiedlichen Therapieansätzen zwischen der aktiven und der passiven Impfung. Bei der aktiven Immunisierung bekommt der Patient Krebsimpfstoffe verabreicht, die in seinem Immunsystem eine Immunantwort auslösen sollen. Die Immunantwort soll dabei idealerweise zum Tod der Tumorzellen oder zumindest zu einem verzögerten Tumorwachstum führen. Im Unterschied dazu erhält der Patient bei der passiven Immunisierung Antikörper oder Antikörper-Fragmente. Diese sollen selektiv an Tumorzellen binden und so zu ihrem Untergang führen. Bei der adoptiven Immuntherapie werden dem Patienten Leukozyten entnommen, ex vivo kultiviert und anschließend wieder dem Patienten injiziert.[2]
Im Bereich der passiven Immunisierung sind bereits mehrere zugelassene Antikörper gegen Krebserkrankungen im klinischen Einsatz. Eine Reihe von Medikamenten zur spezifischen aktiven Immunisierung (Tumorvakzinierung oder Krebsvakzinierung) im Indikationsgebiet der Krebserkrankungen befindet sich noch in der klinischen Entwicklung.
Bei den seit September 2006 in der Europäischen Union zugelassenen HPV-Impfstoffen handelt es sich nicht um eine Krebsimmuntherapie im eigentlichen Sinn. Diese Impfstoffe werden präventiv zur Immunisierung gegen humane Papillomviren (HPV) eingesetzt, die bestimmte Krebsarten – vor allem Gebärmutterhalskrebs – auslösen können.
| Passive Immunisierung | Aktive Immunisierung | ||
|---|---|---|---|
| unspezifisch | spezifisch | unspezifisch | spezifisch |
| Zytokine | Antikörper (mit und ohne Konjugat) | Schlitzschnecken-Hämocyanin | Vakzine aus abgetöteten Tumorzellen |
| Lymphokin-aktivierte Killerzellen oder Zytokin-induzierte Killerzellen |
adoptiver Transfer von T-Lymphozyten | Freund-Adjuvans | Vakzine aus Zellextrakten |
| Peginterferon α | Bacillus Calmette-Guérin | Vakzine auf Basis von Antigenen | |
Einteilung der Krebsimmuntherapie mit Beispielen.[3]
Krebs und das Immunsystem
Immunüberwachung
Die Theorie der Immunüberwachung (engl. immunosurveillance) geht davon aus, dass das Immunsystem nicht nur gegen körperfremde Krankheitserreger, sondern auch gegen körpereigene entartete Zellen aktiv ist.[4] Ein intaktes Immunsystem – so die Schlussfolgerung – ist daher bei einem Organismus ein wichtiges Element zur Vermeidung von Krebserkrankungen.
Welche Auswirkungen ein dauerhaft geschwächtes Immunsystem beim Menschen haben kann, wird an den beiden nachfolgenden Beispielen deutlich.
Das Humane Immundefizienz-Virus (HIV) zerstört CD4+-T-Lymphozyten. Diese zu den weißen Blutkörperchen (Leukozyten) gehörenden Zellen haben eine wichtige Funktion bei der erworbenen Immunantwort. AIDS-Patienten (engl. AIDS = acquired immunodeficiency syndrome = „erworbenes Immundefizienzsyndrom“) haben daher ein geschwächtes Immunsystem. Als eine Folge dieses geschwächten Immunsystems sind verschiedene Krebserkrankungen, wie beispielsweise Lymphome, das Kaposi-Sarkom und das Zervixkarzinom, häufige Komplikationen bei AIDS-Patienten.
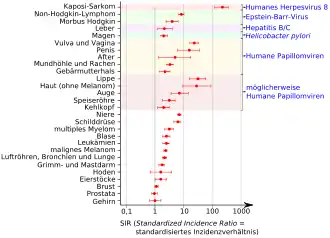
Patienten, die ein solides Spenderorgan (beispielsweise Niere oder Leber) empfangen haben, erhalten – um eine Abstoßung des Fremdorgans zu verhindern – Immunsuppressiva. Dies sind Medikamente, die die Funktionen des Immunsystems vermindern. Statistisch gesehen haben diese Patienten eine um den Faktor drei höhere Wahrscheinlichkeit, an Krebs zu erkranken, als Vergleichsgruppen des gleichen Geschlechts und der gleichen Altersklasse.[5] Nach zehn Jahren Immunsuppression beträgt die Wahrscheinlichkeit, an Krebs zu erkranken, 20 %.[6] Dies gilt vor allem für Krebserkrankungen, die viralen Ursprungs sind, so beispielsweise das Kaposi-Sarkom (Auslöser: Humanes Herpesvirus 8), Morbus Hodgkin und Non-Hodgkin-Lymphom (Epstein-Barr-Virus), Leberkrebs (Hepatitis-C- und Hepatitis-B-Virus), sowie Zervixkarzinom, Vulvakrebs, Vaginalkarzinom, Peniskrebs und andere durch Humane Papillomviren ausgelöste Krebserkrankungen. Aber auch Krebserkrankungen wie kolorektales Karzinom, Nierenkrebs, Blasenkrebs, Schilddrüsenkrebs, multiples Myelom, Leukämie und malignes Melanom – die nach dem gegenwärtigen Kenntnisstand nicht durch Viren ausgelöst werden – nehmen signifikant zu.[5][7] Teilweise wurde nach dem Absetzen der Immunsuppression eine Rückbildung der Malignome, beispielsweise bei malignen Lymphomen, beobachtet.[8][9]
In beiden Gruppen – AIDS-Patienten und Patienten mit Immunsuppressiva nach Transplantationen – wurden hohe Ähnlichkeiten in Bezug auf das Krebsrisiko festgestellt, wobei die Ursache für die erhöhte Krebsrate im Wesentlichen das geschwächte Immunsystem ist.[10]
Die genauen Mechanismen sind noch nicht im Detail verstanden, aber die Immunüberwachung spielt dabei eine entscheidende Rolle.[11]
Die These der Immunüberwachung, das heißt der direkten und ständigen Bekämpfung spontan auftretender Tumoren durch das Immunsystem und der Tumorentstehung als Folge von Schwächen oder Störungen des Immunsystems, wird bis zum heutigen Tag kontrovers diskutiert. Allgemein anerkannt ist dagegen, dass das Immunsystem prinzipiell in der Lage ist, entartete Zellen zu erkennen und erfolgreich zu bekämpfen.[12] Das Immunsystem bietet – wie später noch gezeigt werden wird – keinen vollständigen, sondern einen eher mangelhaften Schutz gegen Tumorerkrankungen, was sich schon an der Tatsache zeigt, dass auch Personen mit einem völlig intakten Immunsystem an Krebs erkranken können. Die Ausnutzung der vorhandenen Mechanismen des Immunsystems sind der Ansatzpunkt für eine Reihe von Strategien zur immunologischen Therapie von Krebserkrankungen.[13]
Immun-Escape


Dass bei völlig gesunden immunkompetenten Menschen das Immunsystem in seiner Abwehrfunktion gegenüber Krebszellen „versagen“ kann, ist eigentlich nicht verwunderlich. Schließlich bilden sich Tumoren aus körpereigenen Zellen der unterschiedlichsten Gewebe, die „außer Kontrolle geraten sind“. Dementsprechend tragen Tumorzellen an ihrer Zelloberfläche das Selbst-Antigen, das sie als „zum Körper gehörend“ ausweist. Dies ist eine völlig andere Situation als bei körperfremden „Eindringlingen“ (Pathogenen), die durch ihre Fremdartigkeit vom Immunsystem relativ leicht zu erkennen sind. Onkologen sprechen daher bei einer Krebserkrankung nicht von einem „Versagen des Immunsystems“.[14] Ein zu scharfes Immunsystem, das auf minimale zelluläre Veränderungen reagieren würde, hätte auf der anderen Seite eine Reihe von Autoimmunerkrankungen zur Folge.
Viele Tumorzellen exprimieren keine mutierten Peptide (tumorspezifische Antigene) an ihrer Oberfläche. Dies wäre eine wesentliche Voraussetzung, dass das Immunsystem die entarteten Zellen als solche erkennen und vernichten kann. Aber auch bei Tumorzellen, die Antigene an ihrer Zellmembran präsentieren, gibt es in vielen Fällen keine ausreichende Immunantwort. Die Ursache ist hier, dass sich die Tumorzellen auf verschiedene Weise dem Immunsystem entziehen können. Man spricht daher vom sogenannten Immun-Escape (engl. immune escape = „Immunentkommen“). So können beispielsweise die auf dem MHC-I-Komplex präsentierten Peptide durch Mutation verändert werden.[15][16][17] Auch die Reduzierung der Expression des Haupthistokompatibilitätskomplexes (MHC) ist beispielsweise eine Reaktion der Krebszellen.[18][19] Ebenso können sich selbige durch die Sekretion von bestimmten immunsuppressiven Zytokinen schützen. Das Antigen-Shedding, das Abwerfen von Antigenen von der äußeren Zellmembran, ist eine Maßnahme der Krebszellen, um der Immunabwehr zu entkommen.[20] Einige Krebszellen sind in der Lage, Fas-Liganden (FasL = Fas-Ligand; CD95) an ihrer Zelloberfläche zu exprimieren. Bindet FasL an den Fas-Rezeptor (FasR), können die Krebszellen bei den FasR-tragenden Lymphozyten den programmierten Zelltod (Apoptose) auslösen. Dieser Vorgang wird als tumor counterattack (dt.: „Tumor-Gegenangriff“) bezeichnet.[1]
Immunoediting
Hinter den Anpassungen der Krebszellen steht keine besondere Strategie oder gar eine eigenständige „Intelligenz“. Sie sind eine Folge von Mutation (durch die Krebszellen selbst) und Selektion (durch das Immunsystem).[19][21] Die am besten angepassten Zellen überleben und vermehren sich weiter (Survival of the Fittest). Die vergleichsweise hohe Proliferations- und Mutationsrate der entarteten Zellen ist dabei für ihre Evolution von Vorteil. Die von den Krebszellen genutzten Abwehrmechanismen sind auch in gesunden Zellen vorhanden, um beispielsweise Autoimmunität zu verhindern.[2] Das Immunsystem bewirkt somit bei Krebszellen zweierlei: zum einen wird der Organismus durch die Zerstörung der entarteten Zellen geschützt und zum anderen wird durch den Selektionsprozess der Tumor „geformt“. Da der Begriff der Immunüberwachung, der nur die Schutzfunktion des Immunsystems betrachtete, dieser erweiterten These nicht mehr ausreichend gerecht wird, spricht man seit Beginn des 21. Jahrhunderts von Immunoediting.[12] Ein Beweis für die These des Immunoediting, also des Selektionsprozesses nicht immunogener Tumorzellen,[22] sind Versuche mit transplantierten Tumoren bei Mäusen. Tumoren, die in immunkompetenten Mäusen mit 3-Methylcholanthren (MCA) erzeugt wurden und somit vom Immunsystem der Maus durch den Selektionsprozess „geformt“ werden konnten, wurden bei der Transplantation in andere Mäuse deutlich weniger oft abgestoßen als Tumoren aus immundefizitären Mäusen. Die Tumoren aus den immundefizitären Mäusen waren durch das eingeschränkte Immunsystem weniger durch Selektionsprozesse geprägt und somit immunogener.[23] Das Immunoediting wird in drei Phasen eingeteilt: Elimination, Gleichgewicht (Equilibrium) und Entkommen (Escape).[12] Die Phase der Elimination entspricht dabei dem ursprünglichen Konzept der Immunüberwachung, in der Krebszellen durch das Immunsystem zerstört werden. In der Gleichgewichtsphase stellt sich nach fast vollständiger Zerstörung der Krebszellen eine immunvermittelte Latenz ein. In der letzten Phase, dem Escape, entkommt der Tumor der Immunüberwachung und wird erst in dieser Phase klinisch sichtbar.[24]
Die Ziele der Krebsimmuntherapie
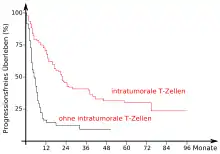
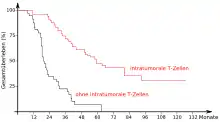
Wie eingangs gezeigt wurde, spielt das Immunsystem bei der Entstehung, beziehungsweise der Vermeidung, von Krebserkrankungen eine wichtige Rolle. Aber auch nachher, wenn durch einen Immun-Escape tatsächlich eine Krebserkrankung ausbricht, hat das Immunsystem einen großen Einfluss auf den Krankheitsverlauf. Dies zeigt sich unter anderem daran, dass die Aktivität des Immunsystems gegenüber Krebszellen ein wichtiger Faktor für die Prognose bei Krebspatienten ist.[26] So konnte beispielsweise gezeigt werden, dass die zellvermittelte Immunogenität gegenüber tumorassoziierten Antigenen (TAA) ein besserer Indikator für die Überlebensrate ist als Staging, Grading und Status der Lymphknoten.[27] Bei Patientinnen mit Brustkrebs in einem frühen Stadium ist die Überlebensrate deutlich höher, wenn sie eine natürliche humorale Immunantwort gegen epitheliales Mucin-1 der Tumorzellen zeigen.[28][29] 2003 wurden in einer Studie über 100 Ovarialkarzinome immunhistologisch untersucht. Dabei konnte eine signifikante Korrelation zwischen dem progressionsfreien Überleben bzw. dem Gesamtüberleben und der Infiltration des Tumorgewebes mit T-Zellen (TIL = tumor infiltrating lymphocytes) festgestellt werden. Die Fünf-Jahres-Überlebensrate betrug bei den Patientinnen mit T-Zell-infiltrierten Tumoren 38 %, während sie bei Patientinnen ohne T-Zellen im Tumor bei lediglich 4,5 % lag.[25] Ähnliche Korrelationen wurden beispielsweise für das maligne Melanom,[30][31] Blasenkrebs,[32] kolorektales Karzinom,[33][34] Prostata,[35] Mastdarmkrebs[36] und Neuroblastom[37] festgestellt.[12]
Die im vorhergehenden Absatz beschriebenen Unzulänglichkeiten und Schwächen des Immunsystems gegenüber Krebszellen sind der Ansatzpunkt der Krebsimmuntherapie. Dabei wurden mehrere, teilweise sehr unterschiedliche, Strategien entwickelt, um direkt oder indirekt über das Immunsystem Krebszellen zu bekämpfen. Das gemeinsame Ziel all dieser Ansätze ist es, Krebszellen mit Hilfe des Immunsystems zu zerstören. Dies kann beispielsweise durch eine unspezifische Stimulierung des Immunsystems, die eine Vermehrung der wichtigsten zytotoxischen Zellen zur Folge hat, geschehen. Auch das „Markieren“ von Tumorzellen mit monoklonalen Antikörpern zum Auslösen einer Immunreaktion[38] ist ein Verfahren der Krebsimmuntherapie, auch wenn die Behandlung mit Antikörpern – nicht nur umgangssprachlich – fälschlicherweise als „Chemotherapie“ bezeichnet wird.
Ungünstigerweise haben die derzeit etablierten Behandlungsschemata – bei allen unbestreitbaren Therapieerfolgen – eine gegenteilige Wirkung auf das Immunsystem: Sowohl chemotherapeutische[39], als auch strahlentherapeutische[40] Maßnahmen schwächen das Immunsystem.[41] Die Proliferation und die Funktion von Lymphozyten ist nach einer Chemotherapie deutlich eingeschränkt. Beispielsweise finden sich im Knochenmark von Patientinnen nach einer adjuvanten (unterstützenden) systemischen Chemotherapie gegen Brustkrebs deutlich weniger T-Zellen, und die Anzahl aktivierter NK-Zellen ist auch über einen längeren Zeitraum reduziert.[26]
Tumorantigene
- → siehe Hauptartikel Tumorantigen
Tumorantigene sind Fragmente von in Tumoren produzierten Proteinen. Diese Fragmente befinden sich auf der äußeren Zellmembran der Tumorzellen, im Zellplasma und im Zellkern. Die Tumorantigene entstehen als Folge des in Krebszellen veränderten Genoms, beziehungsweise durch eine veränderte Genexpression („An- und Ausschalten“ von Genen). Durch diese Veränderungen können neue körperfremde Genprodukte entstehen oder aber Proteine, die beispielsweise normalerweise nur in der embryonalen Entwicklungsphase vorhanden sind. Häufig werden bestimmte – zum Zeitpunkt der Erkrankung auch in gesunden Zellen des Körpers vorhandene – Proteine in großen Mengen produziert (überexprimiert).
Fast alle bisher bekannten Tumorantigene werden über den MHC-I-Komplex auf der Zellmembran präsentiert (Antigenpräsentation).[42] Die dort präsentierten Proteinfragmente bestehen aus ungefähr neun bis zehn Aminosäuren.[43] Die Unterschiede im Aufbau des Antigens oder in der Häufigkeit seiner Expression – im Vergleich zu einer normalen Zelle – machen sie zu potenziellen Zielstrukturen (Targets) für Effektorzellen des Immunsystems[44] und Antikörper[45]. Cytotoxische T-Zellen können über ihren T-Zell-Rezeptor mit Hilfe des CD8-Rezeptor die über den MHC-I-Komplex präsentierten Tumorantigene erkennen und gegebenenfalls die Zielzelle mit dem Tumorantigen zerstören.[43]
Die Tumorantigene sind als Zielstruktur die Basis für die meisten Konzepte der Krebsimmuntherapie.[42] Bisher sind über 2000 Tumorantigene bekannt.[46] Das ideale Tumorantigen – das es in dieser Form nicht gibt – hätte die folgenden Eigenschaften:
- Es wird nur von Krebszellen auf der Zellmembran exprimiert werden. In gesunden Zellen ist es dagegen nicht vorhanden. In solchen Fällen spricht man von einem tumorspezifischen Antigen (TSA).
- Es besteht aus Peptidfragmenten, die über den MHC-I-Komplex an der Zellmembran präsentiert werden und von T-Zellen im MHC-restriktierten Modus erkannt werden.
- Die Expression auf der Zelloberfläche soll über den gesamten Zellzyklus und in möglichst hoher Dichte gewährleistet sein und nicht herunterreguliert werden.
- Das Tumorantigen wird von allen Krebszellen bei allen Krebserkrankungen exprimiert.[43]
In der Realität sieht es anders aus. Die meisten Tumorantigene sind nicht tumorspezifisch (TSA), sondern tumorassoziiert (TAA). Das heißt, dass sie auch von gesunden Zellen exprimiert werden. Allerdings sind bei vielen Tumoren die Tumorantigene überexprimiert. Durch Mutationen im Genom können auch strukturelle Veränderungen in der Proteinsequenz auftreten. Viele Tumorantigene treten nur bei bestimmten Tumorarten und dort oft auch nur in bestimmten Fällen auf. Beispielsweise wird HER2/neu nur bei 20 bis 25 % aller Brustkrebstumoren überexprimiert, weshalb nur in diesen Fällen die Therapie mit dem HER2/neu-spezifischen Antikörper Trastuzumab sinnvoll ist.[47] Zudem können Tumorzellen, beispielsweise als Folge des Immun-Escapes, die Antigenpräsentation über den MHC-I-Komplex herunterfahren.
Die Identifizierung von Tumorantigenen mit hoher Immunogenität, die dem oben gezeigten Idealbild möglichst nahekommen, ist eine der größten Herausforderungen für die Tumorimmunologie und der Schlüssel für erfolgreiche spezifische Immunisierungen.[48] Ein Grundgedanke bei der Identifizierung von Tumorantigenen ist, dass ihre Wahrnehmung durch das Immunsystem ein Indikator für ihre Relevanz in einer Anti-Tumor-Antwort ist.[49] So wurden früher Tumorantigene im Wesentlichen durch die Analyse der Anti-Tumor-Antwort in Patienten identifiziert. Dazu wurden entweder die peripheren[50] oder die tumorinfiltrierenden[51] Lymphozyten (TIL) untersucht oder die Humorale Immunantwort analysiert[52][49] Durch die im Rahmen des Humangenomprojektes gewonnenen Erkenntnisse und mit Hilfe verbesserter analytischer Verfahren hat sich seit einigen Jahren die Reverse Immunologie als leistungsfähiges Hochdurchsatz-Verfahren zur Identifizierung von Tumorantigenen etabliert.[49][53][54][55]
Passive Krebsimmuntherapie
Monoklonale Antikörper
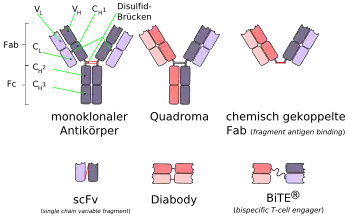
- → siehe Hauptartikel Antikörper und monoklonale Antikörper
Antikörper sind Proteinstrukturen, die mit Hilfe des Schlüssel-Schloss-Prinzips in der Lage sind, Antigene zu erkennen und sich an ihnen festzuheften. Dies trifft auch für Tumorantigene als Zielstrukturen zu. Im menschlichen Körper werden Antikörper von differenzierten B-Lymphozyten, das heißt Plasmablasten und Plasmazellen, produziert. Jeder B-Lymphozyt stellt dabei einen anderen Antikörper her. Sie unterscheiden sich aber nur durch den variablen, zum Antigen-Epitop passenden, Teil – das sogenannte Paratop – am oberen Ende der Y-Struktur. Binden Antikörper über ihr Paratop an ein Antigen-Epitop einer Zelle, so erkennen beispielsweise Makrophagen diesen Immunkomplex und versuchen in der Folge die so markierte Zelle zu vernichten.
Bei der Bekämpfung von Krebszellen spielen die körpereigenen polyklonalen Antikörper allerdings nur eine sehr geringe Rolle. Die meisten Krebszellen präsentieren keine entsprechend stark veränderten Antigene, als dass körpereigene Antikörper in ausreichender Zahl an sie binden könnten. Bei einer höheren Empfindlichkeit der polyklonalen Antikörper würden diese verstärkt auch an gesunde Zellen binden und dort Autoimmunreaktionen auslösen. Monoklonale Antikörper sind dagegen in ihrer Struktur völlig identisch und nur gegen ein Epitop eines Antigens ausgerichtet.[38]
Antikörper spielen vor allem in der Krebsdiagnostik eine wichtige Rolle. Beispielsweise werden bei der Immunszintigrafie monoklonale Antikörper mit Radionukliden markiert, die mittels SPECT im Körper eines Patienten verfolgt werden können (Radiotracer). Reichern sich die so markierten monoklonalen Antikörper in bestimmten Organgeweben an, so kann dies ein Hinweis auf eine Metastasierung sein.
Nach der Zulassung von Rituximab durch die FDA in den Vereinigten Staaten im Jahr 1997 kam der erste therapeutische monoklonale Antikörper (gegen B-Zell-Non-Hodgkin-Lymphome) auf den Markt.[56] Ein Jahr später wurde Trastuzumab gegen metastatisierten Brustkrebs zugelassen.[57] Seitdem wurden weitere monoklonale Antikörper gegen andere Formen von Krebserkrankungen entwickelt und zu deren Behandlung zugelassen.
Die Wirkung monoklonaler Antikörper beruht auf der Bindung an Zielstrukturen, die sich auf der Oberfläche der Zielzelle befinden. Sie können dabei entweder direkt oder indirekt wirken.[58] Bei der direkten Wirkung können die Antikörper durch Kreuzvernetzung des Tumorantigens eine intrazelluläre Signalkaskade in der Krebszelle auslösen. Dies kann eine anti-proliferative Wirkung oder eine unmittelbare Apoptose der Zelle zur Folge haben. Rituximab beispielsweise kann durch die Kreuzvernetzung des Tumorantigens CD20 die Apoptose auslösen.[59]
Die Kreuzvernetzung von Tumorantigenen wie CD22, CD33 oder HLA II mit Antikörpern wirkt dagegen in den Zielzellen anti-proliferativ.[60][61][62][63] Monoklonale Antikörper können auch durch die Blockade von bestimmten Liganden auf Krebszellen wirken. So blockiert Trastuzumab den Her2/neu-Rezeptor, wodurch die Signalkette zum epidermalen Wachstumsfaktor gestört und die Proliferation inhibiert wird. Als Folge davon verlangsamt sich das Tumorwachstum.[64]
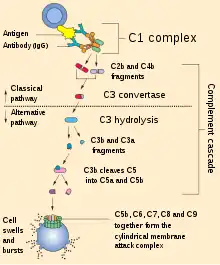
Auf der Fc-Rezeptor-Bindungsstelle (Fc = fragment crystallizable) – dem unteren Teil der Y-Struktur – basieren die indirekten Wirkmechanismen der monoklonalen Antikörper. Damit werden Effektorfunktionen aktiviert. Dazu zählen die komplementabhängige Zytolyse (Complement Dependent Cytolysis, CDC) und die antikörperabhängige zellvermittelte Zytotoxizität (antibody dependent cellular cytotoxicity, ADCC). Bei der CDC wird durch Komplementbindung (complement fixation, CF) die Komplementkaskade ausgelöst, was zur Lyse der Zielzelle führt. Welche Bedeutung, beziehungsweise welchen Anteil, die CDC für den Therapieerfolg hat, ist indes noch unklar.[63][65][66] Bei der ADCC lenken Antikörper über den Fc-Teil zytotoxische Effektorzellen, wie beispielsweise NK-Zellen, die selbst keine Antigenspezifität aufweisen, zur Krebszelle. Die g-Kette des Fc-Teils bewirkt die Signaltransduktion zum Zytoplasma der Effektorzelle. Die Effektorzellen setzen an den Krebszellen lytische Granula, bestehend aus Perforin und Granzyme, frei, die die Apoptose der Krebszellen zur Folge hat. Bei vielen therapeutischen Antikörpern, wie beispielsweise Rituximab, Alemtuzumab, Trastuzumab oder Cetuximab, ist die ADCC der wichtigste Wirkmechanismus.[63] Phagozytierende Zellen wie Makrophagen können ebenfalls über den Fc-Teil eines Antikörpers aktiviert werden und die mit dem Antikörper markierte Zelle phagozytieren.[67] Generell haben die indirekten Wirkmechanismen in der Tumortherapie größere Bedeutung als die direkten.[63][68]
Die therapeutischen Grenzen monoklonaler Antikörper
Eine generelle Problematik für die therapeutische Wirksamkeit der monoklonalen Antikörper ist das Bindungsvermögen der Antikörper an die Krebszellen. Auch bei Krebszellen, die ausreichend Tumorantigene präsentieren, ist die Bindungsrate oft nicht ausreichend hoch. Die direkte Schädigung der Zelle durch assoziierte monoklonale Antikörper ist die seltene Ausnahme. Der Normalfall ist das Auslösen einer Immunreaktion durch den entstandenen Immunkomplex, der – wie im Fall der körpereigenen polyklonalen Antikörper – NK-Zellen, Makrophagen, Lymphozyten und Granulozyten[69] zum Vernichten der so markierten Zellen aktiviert. Für die Therapie größerer solider Tumoren sind monoklonale Antikörper weitgehend ungeeignet. In diesen Fällen liegt ein Verteilungs- und Reichweitenproblem vor. Das Verhältnis von Tumorzellen mit entsprechenden Tumorantigenen zu monoklonalen Antikörpern ist hier zu ungünstig, um ausreichend Tumorzellen zu markieren und durch – ebenfalls in der Anzahl nicht ausreichend vorhandene – Zellen der Immunabwehr abzutöten. Mit einer Molekülmasse von etwa 150 kDa sind Antikörper in ihrer Gewebegängigkeit generell eingeschränkt und können bei soliden Tumoren nur unzureichend in tiefere Schichten eindringen.[70] Antikörperfragmente, wie Fab, F(ab)2 oder scFv, haben hier aufgrund der deutlich geringeren Größe erhebliche Vorteile. Allerdings werden sie auch deutlich schneller aus dem Körper ausgeschieden.[71]
Gegen murine und chimäre Antikörper kann das Immunsystem des Patienten Anti-Antikörper bilden. Bei der Wiederholung eines Therapiezyklusses kann deshalb die Wirksamkeit reduziert sein. Dies trifft selbst auf humanisierte und humane Antikörper zu.[63][68]
Der bereits beschriebene Immun-Escape bewirkt zudem, dass nicht genügend Antikörper den Tumor erreichen. Beispielsweise setzen die Tumorzellen durch das Antigen-Shedding Tumorantigene frei und geben diese in die Blutbahn ab. Dort binden sie an die Antikörper und machen sie so wirkungslos.
Für einige Formen von Krebserkrankungen, wie beispielsweise Leukämien oder Non-Hodgkin-Lymphome, die keine Tumoren bilden, so dass das Konzentrations- und Verteilungsproblem für die monoklonalen Antikörper nicht gegeben ist, werden mit monoklonalen Antikörpern die besten Therapieerfolge erzielt. Im Fall von soliden Tumoren wird die Therapie mit den entsprechenden monoklonalen Antikörpern meist nach operativer Tumorentfernung, Chemotherapie oder Bestrahlung durchgeführt, um einzelne freie Tumorzellen im Körper zu vernichten und dadurch die Bildung von Metastasen zu verhindern.[38]
Bispezifische Antikörper
- → siehe Hauptartikel bispezifischer Antikörper
Antikörperkonjugate
- → siehe Hauptartikel Immunkonjugat
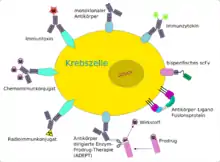
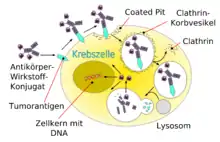
Um die relativ schwache Wirkung der monoklonalen Antikörper bei Krebserkrankungen zu verstärken, wurden verschiedene Strategien des Drug Targetings entwickelt, um die Antikörper als Träger für potentere Wirkstoffe zu nutzen. Der Antikörper dient in diesem Fall als „Transportvehikel“, um den Wirkstoff möglichst spezifisch nur an Tumorzellen zu bringen und diese gezielt abzutöten. Das gesunde Gewebe soll dabei möglichst geschont werden, wodurch die Nebenwirkungen reduziert werden können.[72] Als Wirkstoffe werden Radionuklide[73], Toxine (beispielsweise das Diphtherietoxin), Zytostatika oder auch Zytokine (wie zum Beispiel Interleukin-2) an den entsprechenden Antikörper gebunden (konjugiert). Man spricht in diesen Fällen auch von „bewaffneten Antikörpern“ (engl. armed antibodies).[74][75] Bei Zytokinen spricht man von Immunzytokinen.[76]
Prinzipiell ist es möglich, an Antikörper hochpotente Wirkstoffe zu binden, die bei freier systemischer Gabe aufgrund ihrer hohen Toxizität zu nicht vertretbaren Nebenwirkungen führen würden.[56] Das Konjugat aus Antikörper oder auch Antikörperfragment mit dem Wirkstoff kann als Prodrug angesehen werden, das bei systemischer Gabe eine möglichst geringe Toxizität aufweist und erst am Wirkort, dem Tumor oder auch einer einzelnen Krebszelle, seine volle Wirkung entfaltet.[77]
Die Antikörperkonjugate bestehen aus drei Komponenten:[78]
- dem Antikörper oder Antikörperfragment,
- dem Wirkstoff und
- dem Verbindungsstück (Linker) zwischen Antikörper und Wirkstoff.
- → Für detaillierte Informationen sei auf die jeweiligen Hauptartikel Chemoimmunkonjugat, Immuntoxin und Radioimmuntherapie verwiesen
Aktive Krebsimmuntherapie
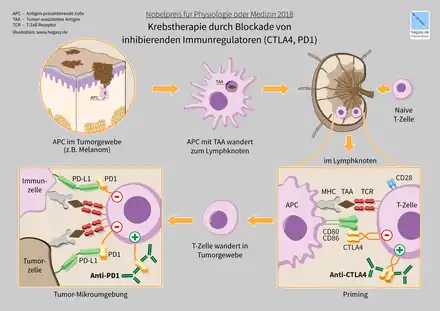
Die aktive Immuntherapie wird üblicherweise noch in die spezifische und in die unspezifische Krebsimmuntherapie unterteilt. Bei der unspezifischen Immuntherapie wird das Immunsystem durch Gabe eines Arzneimittels in seiner Gesamtheit stimuliert. Bei der spezifischen Immuntherapie werden nur bestimmte Zielzellen des Immunsystems angeregt. Ein Beispiel für eine vielversprechende aktive Immuntherapie ist die Verabreichung sogenannter Immun-Checkpoint-Inhibitoren, wie CTLA-4 blockierender Antikörper oder PD-1/PD-L1 spezifischer Antikörper im Melanom. Beide Checkpoint-Inhibitoren führen zu einer gezielten Aktivierung des Immunsystems und erzielten in klinischen Studien erfolgsversprechende Ergebnisse[79].
Schlitzschnecken-Hämocyanin
- → siehe Hauptartikel Schlitzschnecken-Hämocyanin
Schlitzschnecken-Hämocyanin (KLH, engl. Keyhole Limpet Hemocyanin) ist das Sauerstofftransportprotein der Großen Kalifornischen Schlüssellochschnecke (Megathura crenulata). Das Protein dieser in 40 m Wassertiefe lebenden Wasserschnecke aus der Familie der Schlüssellochschnecken (Fissurellidae) basiert auf dem Kupferkomplex Hämocyanin, das bei Säugetieren – deren Sauerstofftransportprotein Hämoglobin auf einem Eisenkomplex aufbaut – eine starke Immunantwort auslöst. Es ist unter dem internationalen Freinamen Immunocyanin (Markenname Immucothel) in den Niederlanden, Österreich und Südkorea,[80] aber nicht in Deutschland, zur Redizivprophylaxe bei Blasenkrebs nach transurethraler Resektion zugelassen.
Mitte der 1960er Jahre wurde die immunstimulierende Wirkung von Hämocyanin bei Säugetieren erstmals festgestellt.[81][82][83] Ab 1967 wurde KLH dann beim Menschen für diagnostische Zwecke in Immunkompetenztests verwendet.[84] Auch der US-amerikanische Urologe Carl A. Olsson verwendete KLH für immundiagnostische Zwecke bei Patienten mit oberflächlichem Blasenkrebs. In der Folge bemerkte er, dass die Patienten, die getestet wurden, signifikant weniger Rezidive entwickelten als die nicht getesteten Patienten.[85][86]
Die immunstimulierende Wirkung von KLH lässt sich durch die Sekundärstruktur von Hämocyanin erklären, die teilweise der von Antikörpern, Histokompatibilitätsmolekülen oder Bence-Jones-Proteinen gleicht.[87] Hinzu kommen epitopartige Sequenzen auf der Oberfläche des Proteins.[86]
KLH wird auch als Träger für Haptene verwendet.[88] Dabei werden nicht immunogene Epitope mit dem immunogenen KLH zu einem Immunkonjugat verknüpft, um so eine immunogene Wirkung auf das Epitop zu erreichen.[89] Die Moleküle vieler Antigen-Epitope sind zu klein, um vom Immunsystem erkannt zu werden. Mit dieser Methode können auch Antigene dem Immunsystem als „feindlich“ präsentiert werden, die der Körper sonst toleriert. Bei großen Trägermolekülen wie KLH können auch mehrere unterschiedliche Antigene an die Oberfläche des Trägers gebunden werden.[80]
Bacillus Calmette-Guérin
- → siehe Hauptartikel Bacillus Calmette-Guérin
Das Bacillus Calmette-Guérin (BCG) wurde zu Beginn des 20. Jahrhunderts aus Rindertuberkelbazillen gewonnen und als Lebendimpfstoff gegen Tuberkulose eingesetzt. 1959 wurde bei Mäusen mit transplantierten Tumoren erstmals ein positiver Effekt bezüglich der Rückbildung der Tumoren bei der Infektion mit BCG festgestellt. Die Infektion erhöhte die Aktivität des retikulohistiozytären Systems und die Zahl der Antikörper in den Versuchstieren.[90]
Bei der direkten Injektion in Tumoren konnte in vielen Fällen eine Rückbildung des Tumors beobachtet werden. Vielversprechend verliefen die Behandlungsversuche bei metastatisierten Melanomen, wo die systemische Gabe von BCG auch zu einer Rückbildung der Metastasen führte[91] und die Überlebensrate der so behandelten Patienten signifikant erhöht wurde.[92] In randomisierten, kontrollierten Studien konnten die positiven Resultate allerdings nicht bestätigt werden.[93][94]
1976 wurden erstmals positive Ergebnisse bei der Behandlung des oberflächlichen Blasenkrebses mit BCG veröffentlicht.[95] Dabei wurde BCG direkt in die Harnblase injiziert (intravesikal).[96] In einer Vielzahl von klinischen Studien konnte die therapeutische Wirksamkeit bei der Behandlung von oberflächlichem Blasenkrebs – hierbei ist der Tumor auf die innere Auskleidung der Harnblase beschränkt – nachgewiesen werden.[97][98][99] Die Therapie mit BCG ist bei dieser Erkrankung der Goldstandard.[100][101][102] Nach der Einschätzung mehrerer Autoren ist dies bis heute die erfolgreichste Krebsimmuntherapie.[101][103] BCG ist dabei jedem Chemotherapeutikum deutlich überlegen. Die Wahrscheinlichkeit für ein Tumorrezidiv ist – im Vergleich zur intravesikalen Chemotherapie – nur halb so hoch. In über 80 % der Fälle wird eine Eradikation, das heißt eine vollständige Eliminierung des Tumors, erreicht.[104]
Der genaue Wirkungsmechanismus von BCG ist noch nicht aufgeklärt. Die Blase ist ein weitgehend abgeschottetes und abgeschlossenes Organ, in dem BCG eine sehr hohe lokale Konzentration erreichen und eine komplexe lang anhaltende lokale Aktivierung des Immunsystems bewirken kann.[103] Durch die lokale Aktivierung werden verschiedene Zytokine in den Urin,[105][106] beziehungsweise an das Blasengewebe, abgegeben. Gleichzeitig infiltrieren Granulozyten und Monozyten die Blasenwand.[107][108] Dendritische Zellen schütten TNF-α und Interleukin-12 (IL-12) aus. Die Ausschüttung dieser Zytokine aktiviert wiederum Zellen der angeborenen Immunabwehr (STIL), wodurch sich IL-12-Rezeptor-exprimierende NK-Zellen stark vermehren (proliferieren).[109]
Die wichtigsten Nebenwirkungen bei der Therapie mit BCG sind: Zystitis (Entzündung der Harnblase), Pollakisurie (häufiger Harndrang), Hämaturie (Blut im Harn) und Fieber. Bei Studien mit zusammen über 500 Patienten ist kein durch BCG bedingter Todesfall bekannt.[110]
Adoptiver Zelltransfer
Tumoren können auch mithilfe eines adoptiven Zelltransfers behandelt werden. Für diese Therapie werden beispielsweise autologe (also vom Patienten selbst stammende) T-Zellen verwendet, welche mit einem tumorantigenspezifischen T-Zellrezeptor[111] oder chimeren Antigenrezeptor (CAR)[112] ausgestattet wurden. Hierdurch erhalten die T-Zellen eine neue, gegen den Tumor gerichtete Spezifität. Präsentieren die Tumorzellen die dem Rezeptor entsprechenden Antigene auf ihrer Oberfläche, können sie durch die T-Zellen eliminiert werden. Auch die Therapie mit Tumor-infiltrierenden Lymphozyten (TILs)[113] oder mit antigenbeladenen dendritischen Zellen[114] ist möglich. Letztere dient der In-vivo-Induzierung einer Immunantwort und der Aktivierung tumorantigenspezifischer T-Zellen im Körper der Patienten.[115]
Geschichte

Die Idee, das Immunsystem so zu beeinflussen, dass es in der Lage ist, Neoplasien erkennen und zerstören zu können, ist schon sehr alt. Die ersten Berichte über Tumorregressionen nach Infektionskrankheiten datieren auf das Jahr 1550 vor Christus.[116] Im Papyrus Ebers war die empfohlene Behandlung von Schwellungen (Tumoren) ein Kataplasma (eine Art Breiwickel, in dem sich ein Gemisch aus Pflanzenpulvern, Samen und anderen Arzneistoffen befand), mit anschließendem Einschnitt in den Tumor.[117] Eine solche Behandlung führt zu einer Infektion des Tumors.[118]
Die ersten immunologischen Versuche der Neuzeit führte 1777 James Nooth, Arzt von Edward Augustus, Duke of Kent and Strathearn, und Mitglied des Royal Collage of Surgeons, durch. Nooth implantierte sich mehrmals fremdes Tumorgewebe in kleine Einschnitte seines Armes, um eine Krebsprophylaxe zu erreichen. Die Implantierung hatte lediglich Entzündungsreaktionen und leichtere Schmerzen zur Folge. Ein ähnliches Ergebnis erhielt 1808 Jean-Louis Alibert, der Leibarzt von König Ludwig XVIII., der sich von einem Kollegen Flüssigkeit einer Brustkrebspatientin injizieren ließ. Es stellte sich dabei lediglich eine Entzündungsreaktion ein.[119][120]


Deutlich erfolgreicher waren dagegen die Experimente des US-Amerikaners William Coley (1862–1936), der als Pionier der Krebsimmuntherapie gilt. Coley war Arzt am Memorial Sloan-Kettering Cancer Center in New York City. Er hatte von einem Krebspatienten gehört, bei dem es zu einer vollständigen Remission nach hohem Fieber, bedingt durch ein Erysipel (Wundrose, eine bakterielle Infektion der oberen Hautschichten und Lymphwege), kam. Coley stellte fest, dass auch Robert Koch, Louis Pasteur, Paul von Bruns und Emil von Behring ähnliche Fälle nach einem Erysipel beschrieben hatten. So veröffentlichte Bruns 1888[121] zusammenfassend seine klinischen Beobachtungen über vollständige Sarkom-Rückbildungen bei spontanem oder auch künstlich herbeigeführten Erysipel.[122] 1891 injizierte Coley einem Krebspatienten – einem 40-jährigen italienischen Immigranten, der bereits zwei Operationen nach Rezidiven hinter sich und nach der Prognose nur noch wenige Wochen vor sich hatte – die Erysipel-Bakterien der Art Streptococcus pyogenes direkt in den Tumor.[123] Coley wiederholte die Injektionen über mehrere Monate, und der Tumor bildete sich bei dem Patienten zurück.[124] Der Patient überlebte acht Jahre.[125][126][127][128] Später verwendete Coley eine Mischung (Coley’s Toxin) abgetöteter Bakterien der Arten Streptococcus pyogenes und Serratia marcescens,[129] zusammen mit den noch aktiven Endotoxinen, direkt in Tumoren. Bei Weichteilsarkomen erreichte Coley mit seiner Methode die beachtliche Heilungsrate von 10 %.[120] Die Ansprechraten waren sehr unterschiedlich und die Nebenwirkungen beachtlich. Ab 1899 produzierte die Parke-Davis Corporation Coley’s Toxin, das die folgenden 30 Jahre eine weite Verbreitung erfuhr.[130][131] Ein wesentlicher Grund für die Verbreitung war, dass es bis 1934 die einzige systemische Krebstherapie war.[132] Mit der Entwicklung der Strahlentherapie und Fortschritten bei der Chemotherapie geriet Coley’s Toxin weitgehend in Vergessenheit.[118][133] Parke-Davis stellte 1952 die Produktion von Coley’s Toxin ein,[130] und die FDA verweigerte 1962 die Anerkennung als geprüften Wirkstoff.[118] Der genaue Wirkungsmechanismus ist bis zum heutigen Tag noch ungeklärt, beruht aber sehr wahrscheinlich auf dem Auslösen einer Zytokin-Kaskade, die eine spezifische und unspezifische Immunantwort zur Folge hat.[94]

Paul Ehrlich formulierte als Erster 1909 die These, dass das Immunsystem Tumorzellen erkennen und beseitigen kann. Auf diese Weise würden viele Tumoren schon in einem sehr frühen Anfangsstadium eliminiert und der Körper so vor einer deutlich höheren Inzidenz an malignen Tumoren geschützt werden.[134]
„Ich bin überzeugt, dass aberrierende Keime bei dem kolossal komplizierten Verlauf der fötalen und post-fötalen Entwicklung außerordentlich häufig vorkommen, daß sie aber glücklicherweise bei der überwiegenden Mehrzahl der Menschen vollkommen latent bleiben, dank der Schutzvorrichtungen des Organismus. Würden diese nicht bestehen, so könnte man vermuten, daß das Karzinom in einer geradezu ungeheuerlichen Frequenz auftreten würde“
Der US-amerikanische Biostatistiker Raymond Pearl veröffentlichte 1929 eine Autopsiestudie, in der er eine signifikant niedrigere Krebsrate bei Patienten mit Tuberkulose feststellte.[136] Als sich im Jahr darauf das Lübecker Impfunglück mit dem Bacillus Calmette-Guérin (BCG) – einem Impfstoff gegen Tuberkulose – ereignete, bedeutete dies das vorläufige Aus für entsprechende Therapieansätze in der Onkologie. Erst gegen Ende der 1950er Jahre griff Lloyd J. Old die Idee wieder auf und führte BCG erfolgreich in die Krebsimmuntherapie ein. Ab 1969 wurde BCG dann in der klinischen Praxis eingesetzt.[101]

Lewis Thomas (1913–1993)[137] und der spätere Nobelpreisträger Frank Macfarlane Burnet[138] griffen unabhängig voneinander Ehrlichs These in den 1950er beziehungsweise 1970er Jahren wieder auf und formulierten die Hypothese der „Immunüberwachung“ (immunosurveillance) bei der Tumorentstehung.[139] Sowohl Thomas als auch Burnet vermuteten, dass die T-Zellen eine wesentliche Rolle bei der immunologischen Überwachung spielen.[140]
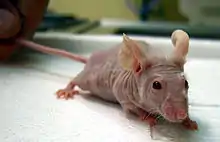
Die Hypothese der Immunologischen Überwachung war lange Zeit sehr umstritten und schien durch Versuche mit Nacktmäusen in den 1970er Jahren sogar widerlegt zu sein.[141] Nacktmäuse, auch athymische Mäuse genannt, haben durch den fehlenden Thymus ein stark eingeschränktes Immunsystem. Der These der Immunüberwachung entsprechend, sollten diese Mäuse gegenüber Mäusen des Wildtyps erheblich häufiger Tumoren entwickeln. Bei vergleichenden Versuchen mit 3-Methylcholanthren – einem starken Karzinogen, das bei Mäusen Sarkome induziert – war in der Tumorrate kein signifikanter Unterschied zwischen athymischen und normalen Mäusen zu erkennen.[142] Einige Jahre später wurde allerdings festgestellt, dass Nacktmäuse entgegen den ersten Vermutungen nicht völlig frei von T-Zellen sind,[143][144] und zudem über eine normale Anzahl von NK-Zellen verfügen.[145] Speziell die NK-Zellen können von sich aus gegen Tumorzellen aktiv werden.[146]
Der definitive Beweis für die Immunüberwachung wurde 2001 durch Vijay Shankaran und Kollegen erbracht. Knockout-Mäuse, bei denen das RAG2- und das STAT1-Gen abgeschaltet wurden, hatten eine erheblich erhöhte Inzidenz zur spontanen oder chemisch induzierten Tumorbildung. Die Genprodukte von RAG2 und STAT1 spielen für die Reifung der T- und B-Zellen, beziehungsweise bei der Signalvermittlung nach Bindung an den γ-Interferon-Rezeptor, eine wichtige Rolle.[23][42][147]
Pierre van der Bruggen und Kollegen identifizierten 1991 am Ludwig Institute for Cancer Research in Brüssel mit MAGEA1 (melanoma antigen family A, 1) erstmals ein Epitop malignen Ursprungs, das von T-Zellen erkannt wird.[50] In der Folge wurden – unter anderem mit der cDNA-Expressionsklonierung – über 70 weitere tumorassoziierte Antigene mit immunogenen Eigenschaften gefunden.[42]
Die beiden amerikanischen Immuntherapeuten James P. Allison und Carl H. June werden im März 2015 für ihre bahnbrechenden Arbeiten zur Immuntherapie gegen Krebs mit dem 100.000 € dotierten Paul Ehrlich- und Ludwig Darmstaedter-Preis ausgezeichnet. James Allison ist Pionier der Checkpoint-Hemmung zur Behandlung fortgeschrittener Melanome, Carl June hat die CART-19-Therapie gegen Leukämie entwickelt.[148]
Siehe auch
Literatur
- Deutsches Krebsforschungszentrum (Hrsg.): Einblick: Waffen des Immunsystems. (PDF; 3,7 MB) Ausgabe 1/2010
- C. Huber (Hrsg.) u. a.: Krebsimmuntherapien. Deutscher Ärzteverlag, 2007, ISBN 3-7691-1212-1
- G. C. Prendergast und E. M. Jaffee: Cancer Immunotherapy. Academic Press, 2007, ISBN 0-12-372551-8 (englisch)
- J. P. Allison u. a.: Cancer Immunotherapy. Academic Press, 2006, ISBN 0-12-022489-5 (englisch)
- M. L. Disis: Immunotherapy of cancer. Humana Press, 2006, ISBN 1-58829-564-8 (englisch)
- J. H. Finke, R. M. Bukowski: Cancer immunotherapy at the crossroads. Humana Press, 2004, ISBN 1-58829-183-9 (englisch)
- P. L. Stern u. a.: Cancer vaccines and immunotherapy. Cambridge University Press, 2000, ISBN 0-521-62263-8 (englisch)
Einzelnachweise
- S. Lang: Rekombinante Parvoviren in der Gentherapie von Krebs: Vektorcharakterisierung und Analyse der Wirksamkeit. Dissertation, Ruprecht-Karls-Universität Heidelberg, 2003.
- F. O. Losch: Aktivierung von T-Lymphozyten durch melanomspezifische variante Antigen-Rezeptoren. Dissertation, Albert-Ludwigs-Universität Freiburg, 2001
- Tumorimmunologie. (Memento des Originals vom 22. März 2005 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. (PDF; 1,9 MB) Institut für Immunologie der Universität Kiel
- W. G. Land: Immunsuppressive Therapie. Georg Thieme Verlag, 2006, ISBN 3-13-133621-8, S. 134.
- C. M. Vajdic und M. T. van Leeuwen: Cancer incidence and risk factors after solid organ transplantation. In: Int J Cancer 2009, [Epub ahead of print]. PMID 19444916
- A. Kapoor: Malignancy in kidney transplant recipients. In: Drugs 68, 2008, S. 11–19. PMID 18442297 (Review)
- A. Gutierrez-Dalmau und J. M. Campistol: Immunosuppressive therapy and malignancy in organ transplant recipients: a systematic review. In: Drugs 67, 2007, S. 1167–1198. PMID 17521218 (Review)
- T. E. Starzl u. a.: Reversibility of lymphomas and lymphoproliferative lesions developing under cyclosporin-steroid therapy. In: The Lancet 8377, 1984, S. 583–587. PMID 6142304
- A. M. Asemissen: Phänotyp-Charakterisierung reaktiver T-Zellen von Melanompatienten gegen autologe Tumorzellen und ein neues Tyrosinase-Epitop. Dissertation, Humboldt-Universität zu Berlin, 2004
- A. E. Grulich: Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. In: The Lancet 370, 2007, S. 59–67. PMID 17617273
- C. M. Vajdic und M. T. van Leeuwen: What types of cancers are associated with immune suppression in HIV? Lessons from solid organ transplant recipients. In: Curr Opin HIV AIDS 4, 2009, S. 35–41. PMID 19343829 (Review)
- G. P. Dunn u. a.: Cancer immunoediting: from immunosurveillance to tumor escape. In: Nat Immunol 3, 2002, S. 991–998. PMID 12407406 (Review)
- Tumorspezifisches Targeting der humanen Natürlichen Killerzellinie YT durch Gentransfer chimärer Immunglobulin-Tzellrezeptoren. Dissertation, Humboldt-Universität zu Berlin, 2005.
- Das Immunsystem: Funktion und Bedeutung bei Krebs, Krebsinformationsdienst des Deutschen Krebsforschungszentrums (DKFZ), Heidelberg. 28. September 2010. Zuletzt abgerufen am 4. September 2014.
- P. G. Coulie u. a.: A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. In: PNAS 92, 1995, S. 7976–7980. PMID 7644523
- P. F. Robbins u. a.: A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. In: J Exp Med 183, 1996, S. 1185–1192. PMID 8642260
- T. Wolfel u. a.: A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. In: Science 269, 1995, S. 1281–1284. PMID 7652577
- E. P. Cohen und T. S. Kim: Neoplastic cells that express low levels of MHC class I determinants escape host immunity. In: Semin Cancer Biol 5, 1994, S. 419–428. PMID 7703441 (Review)
- I. Algarra u. a.: The selection of tumor variants with altered expression of classical and nonclassical MHC class I molecules: implications for tumor immune escape. In: Cancer Immunol Immunother 53, 2004, S. 904–910. PMID 15069585 (Review)
- P. H. Black: Shedding from the cell surface of normal and cancer cells. In: Adv Cancer Res 32, 1980, S. 75–199. PMID 7008543 (Review)
- H. T. Khong und N. P. Restifo: Natural selection of tumor variants in the generation of "tumor escape" phenotypes. In: Nat Immunol 3, 2002, S. 999–1005. PMID 12407407 (Review)
- U. Gnad-Vogt u. a.: Pankreaskarzinom: EGFR- und Immuntherapie. In: J Onkologie 4, 2006
- V. Shankaran u. a.: IFNγ prevent prevent primary tumour development and shape tumour immunogenicity. In: Nature 410, 2001, S. 1107–1111. PMID 11323675
- D. Nargosen: Antigen-spezifische T-Zell-Immunität und Antigen-präsentierende Zellen bei malignen Erkrankungen. Habilitationsschrift, Charité Berlin, 2006
- L. Zhang u. a.: Intratumoral T Cells, Recurrence, and Survival in Epithelial Ovarian Cancer. In: NEJM 348, 2003, S. 203–213. PMID 12529460
- E. F. Solomayer u. a.: Influence of adjuvant hormone therapy and chemotherapy on the immune system analysed in the bone marrow of patients with breast cancer. In: Clin Cancer Res 9, 2003, S. 174–180. PMID 12538466
- J. L. Mccoy u. a.: Cell-mediated immunity to tumor-associated antigens is a better predictor of survival in early stage breast cancer than stage, grade, or lymph node status. In: Breast Cancer Res 60, 2000, S. 227–234. PMID 10930110
- S. Von Mensdorrf-Pouilly u. a.: Survival in early breast cancer patients is favourably influenced by a natural humoral immune response to polymorphic epithelial mucin. In: J Clin Oncol 18, 2000, S. 547–582. PMID 10653872
- F. Rilke u. a.: Prognostic significance of HER-2/neu expression in breast cancer and its relationship to other prognostic factors. In: J Cancer 49, 1991, S. 44–49. PMID 1678734
- W. H. Clark u. a.: Model predicting survival in stage I melanoma based on tumor progression. In: J Natl Cancer Inst 81, 1989, S. 1893–1904. PMID 2593166
- C. G. Clemente u. a.: Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. In: Cancer 77, 1996, S. 1303–1310. PMID 8608507
- P. K. Lipponen u. a.: Tumour infiltrating lymphocytes as an independent prognostic factor in transitional cell bladder cancer. In: Eur J Cancer 29A, 1992, S. 69–75. PMID 1445749
- L. Nacopoulou u. a.: Prognostic significance of histologic host response in cancer of the large bowel. In: Cancer 47, 1981, S. 930–936. PMID 7226044
- Y. Naito u. a.: CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. In: Cancer Res 58, 1998, S. 3491–3494. PMID 9721846
- N. A. Epstein und L. P. Fatti: Prostatic carcinoma: some morphological features affecting prognosis. In: Cancer 37, 1976, S. 2455–2465. PMID 1260728
- J. R. Jass: Lymphocytic infiltration and survival in rectal cancer. In: J Clin Pathol 39, 1986, S. 585–589. PMID 3722412
- L. Palma u. a.: Lymphocytic infiltrates in primary glioblastomas and recidivous gliomas. Incidence, fate, and relevance to prognosis in 228 operated cases. In: J Neurosurg 49, 1978, S. 854–861. PMID 731302
- Monoklonale Antikörper: Wichtig in Diagnostik und Therapie, Krebsinformationsdienst des Deutschen Krebsforschungszentrums (DKFZ), Heidelberg. Vom 27. Juni 2014. Zuletzt abgerufen am 4. September 2014.
- D. H. Kang u. a.: Significant impairment in immune recovery after cancer treatment. In: Nurs Res 58, 2009, S. 105–114. PMID 19289931
- S. Uh u. a.: The effect of radiation therapy on immune function in patients with squamous cell lung carcinoma. In: Chest 105, 1994, S. 132–137. PMID 7903922
- M. Tagawa u. a.: Virology- and immunology-based gene therapy for cancer. In: Cancer Immunol Immunother 55, 2006, S. 1420–1425. PMID 16691360 (Review)
- K. Kokowski: Zelluläre Immunantwort gegen die Tumorantigene Muzin und Telomerase bei Patienten mit Mammakarzinom. Dissertation, FU Berlin, 2008.
- R. H. Vonderheide: Telomerase as a universal tumor-associated antigen for cancer immunotherapy. In: Oncogene 21, 2002, S. 674–679. PMID 11850795 (Review)
- B. J. van den Eynde und P. van der Bruggen: T cell defined tumor antigens. In: Curr Opin Immunol9, 1997, S. 684–693. PMID 9368778 (Review)
- L. M. Weiner u. a.: Monoclonal antibodies for cancer immunotherapy. In: Lancet 373, 2009, S. 1033–1040. PMID 19304016 (Review)
- F. Yang und X. F. Yang: New concepts in tumor antigens: their significance in future immunotherapies for tumors. (PDF; 251 kB) In: Cell Mol Immunol 2, 2005, S. 331–341. PMID 16368059 (Review)
- M. Untch u. a.: Adjuvante Therapie mit Trastuzumab bei Mammakarzinompatientinnen. In: Dtsch Arztebl 103, 2006, S. A-3406/B-2961/C-2841
- S. Viatte u. a.: Reverse immunology approach for the identification of CD8 T-cell-defined antigens: Advantages and hurdles. In: Immunology and Cell Biology 84, 2006, S. 318–330. PMID 16681829 doi:10.1111/j.1440-1711.2006.01447.x (Review)
- J. D. Gordan und R. H. Vonderheide: Universal tumor antigens as targets for immunotherapy. In: Cytotherapy 4, 2002, S. 317–327. PMID 12396831 (Review)
- P. van der Bruggen u. a.: A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. In: Science 254, 1991, S. 1643–1647. PMID 1840703
- Y. Kawakami u. a.: Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2-restricted tumor infiltrating lymphocytes. In: J Exp Med 180, 1994, S. 347–352. PMID 7516411
- U. Sahin u. a.: Serological identification of human tumor antigens. In: Curr Opin Immunol 9, 1997, S. 709–716. PMID 9368781 (Review)
- M. Schirle u. a.: Identification of tumor-associated MHC class I ligands by a novel T cell-independent approach. In: Eur J Immunol 30, 2000, S. 2216–2225. PMID 10940913
- M. Schwarz: Identifizierung potentiell immunogener Peptide auf Zellen der chronischen myeloischen Leukämie. Dissertation, FU Berlin, 2004
- JC Castle et al.: Exploiting the mutanome for tumor vaccination. In: Cancer Research 2(5):1081-91. PMID 22237626
- D. Schrama u. a.: Antibody targeted drugs as cancer therapeutics. In: Nat Rev Drug Discov 5, 2006, S. 147–159. PMID 16424916
- S. Rueckert u. a.: A monoclonal antibody as an effective therapeutic agent in breast cancer: trastuzumab. In: Expert Opinion on Biological Therapy 5, 2005, S. 853–866. PMID 15952915
- M. J. Glennie und P. W. Johnson: Clinical trials of antibody therapy. In: Immunol Today 21, 2000, S. 403–410. PMID 10916144 (Review)
- D. Shan u. a.: Apoptosis of malignant human B cells by ligation of CD20 with monoclonal antibodies. In: Blood 91, 1998, S. 1644–1652. PMID 9473230
- M. Dechant u. a.: HLA class II antibodies in the treatment of hematologic malignancies. In: Semin Oncol 30, 2003, S. 465–475. PMID 12939715 (Review)
- R. Meng u. a.: The evaluation of recombinant, chimeric, tetravalent antihuman CD22 antibodies. In: Clin Cancer Res 10, 2004, S. 1274–1281. PMID 14977825
- C. Vitale u. a.: Engagement of p75/AIRM1 or CD33 inhibits the proliferation of normal or leukemic myeloid cells. In: PNAS 96, 1999, S. 15091–15096. PMID 10611343
- C. Kellner: Entwicklung und Charakterisierung bispezifischer Antikörper-Derivate zur Immuntherapie CD19-positiver Leukämien und Lymphome. Dissertation, Friedrich-Alexander-Universität Erlangen-Nürnberg, 2008.
- M. Harries und I. Smith: The development and clinical use of trastuzumab (Herceptin). (Memento des Originals vom 2. Dezember 2009 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. In: Endocr-Relat Cancer 9, 2002, S. 75–85. PMID 12121832 (Review)
- O. Manches u. a.: In vitro mechanisms of action of rituximab on primary non-Hodgkin lymphomas. In: Blood 101, 2003, S. 949–954. PMID 12393572
- W. K. Weng und R. Levy: Expression of complement inhibitors CD46, CD55, and CD59 on tumor cells does not predict clinical outcome after rituximab treatment in follicular non-Hodgkin lymphoma. In: Blood 98, 2001, S. 1352–1357. PMID 11520782
- O. H. Brekke und I. Sandlie: Therapeutic antibodies for human diseases at the dawn of the twenty-first century. In: Nat Rev Drug Discov 2, 2003, S. 52–62. PMID 12509759 (Review)
- P. Carter: Improving the efficacy of antibody-based cancer therapies. In: Nat Rev Cancer 1, 2001, S. 118–129. PMID 11905803 (Review)
- I. Heisler: Bedeutung spaltbarer Peptidlinker für die Funktion rekombinanter Saporin-EGF-Immunotoxine. Dissertation, FU Berlin, 2008
- R. K. Jain und L. T. Baxter: Mechanisms of heterogeneous distribution of monoclonal antibodies and other macromolecules in tumors: significance of elevated interstitial pressure. In: Cancer Res 48, 1988, S. 7022–7032. PMID 3191477
- T. Yokota u. a.: Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. In: Cancer Res 52, 1992, S. 3402–3408. PMID 1596900
- J. Chen u. a.: Antibody-cytotoxic agent conjugates for cancer therapy. In: Expert Opin Drug Deliv 2, 2005, S. 873–890. PMID 16296784 (Review)
- D. E. Milenic u. a.: Antibody-targeted radiation cancer therapy. In: Nat Rev Drug Discov 3, 2004, S. 488–499. PMID 15173838
- D. Baron: Therapeutischer Einsatz monoklonaler Antikörper. In: Zeitschrift Naturwissenschaften 84, 1997, S. 189–198. doi:10.1007/s001140050376
- G. C. MacDonald und N. Glover: Effective tumor targeting: strategies for the delivery of Armed Antibodies. In: Curr Opin Drug Discov Devel 8, 2005, S. 177–183. PMID 15782542 (Review)
- R. A. Reisfeld u. a.: Immunocytokines: a new approach to immunotherapy of melanoma. In: Melanoma Res 7, 1997, S. 99–106. PMID 9578424 (Review)
- S. Jaracz u. a.: Recent advances in tumor-targeting anticancer drug conjugates. In: Bioorg. Med. Chem. 13, 2005, S. 5043–5054. PMID 15955702 (Review)
- R. V. Chari: Targeted cancer therapy: conferring specificity to cytotoxic drugs. In: Acc Chem Res 41, 2008, S. 98–107. PMID 17705444 (Review)
- Semin Oncol. 2015 Jun;42(3):363–377. doi:10.1053/j.seminoncol.2015.02.015. Epub 2015 Feb 16. Immune Checkpoint Protein Inhibition for Cancer: Preclinical Justification for CTLA-4 and PD-1 Blockade and New Combinations. Baksh K1, Weber J2.
- B. Pfeifer u. a.: Onkologie integrativ. Verlag Elsevier, Urban&Fischer, 2006, ISBN 3-437-56420-X, S. 239.
- W. O. Weigle: Immunochemical properties of hemocyanin. In: Immunochem 1, 1964, S. 295–302. PMID 14250783
- J. E. Curtis u. a.: The human primary immune response to keyhole limpet hemocyanin: interrelationships of delayed hypersensitivity, antibody response and in vitro blast transformation. In: Clin Exp Immunol 6, 1970, S. 473–491. PMID 4320164
- F. J. Dixon u. a.: The antibody responses of rabbits and rats to hemocyanin. In: J Immunol 97, 1966, S. 350–355. PMID 5925706
- M. A. Swanson und R. S. Schwartz: Immunosuppressive therapy. The relation between clinical response and immunologic competence. In: NEJM 277, 1967, S. 163–170. PMID 4166031
- C. A. Olsson, R. Chute, C. N. Rao: Immunologic reduction of bladder cancer recurrence rate. In: The Journal of urology. Band 111, Nummer 2, Februar 1974, S. 173–176, PMID 4810758.
- J. Schütz: Isolierung, Sequenzierung und Untersuchung physikalisch-chemischer Eigenschaften von strukturellen Untereinheiten und funktionellen Einheiten verschiedener Arthropoden- und Molluskenhämocyanine. Dissertation, Eberhard-Karls-Universität Tübingen, 2000
- R. Roth: Hämocyanin – ein starkes Antigen In: Immunologie Spektrum 3, Boehringer GmbH, Mannheim, 1990.
- J. Stoschek: biosyn Arzneimittel: Integratives Konzept in der Onkologie als Ziel. In: Dtsch Arztebl 98, 2001, S. A-1983/B-1678/C-1491.
- C. Huber u. a. (Herausgeber) Krebsimmuntherapien. Deutscher Ärzteverlag, 2007, ISBN 3-7691-1212-1, S. 160.
- L. J. Old u. a.: Effect of Bacillus Calmette-Guerin infection on transplanted tumours in the mouse. In: Nature 184, 1959, S. 291–292. PMID 14428599
- D. Morton u. a.: Immunological factors which influence response to immunotherapy in malignant melanoma. In: Surgery 68, 1970, S. 158–163. PMID 10483463
- F. R. Eilber u. a.: Results of BCG adjuvant immunotherapy for melanoma of the head and neck. In: Am J Surg 132, 1976, S. 476–479. PMID 1015538
- R. Molife und B. W. Hancock: Adjuvant therapy of malignant melanoma. In: Crit Rev Oncol Hematol 44, 2002, S. 81–102. PMID 12399001 (Review)
- I. D. Davis u. a.: Rational approaches to human cancer immunotherapy. (Memento des Originals vom 11. April 2009 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. In: J Leukoc Biol 73, 2003, S. 3–29. PMID 12525559 (Review)
- A. Morales u. a.: Intracavitary Bacillus Calmette-Guerin in the treatment of superficial bladder tumors. In: The Journal of Urology 116, 1976, S. 180–183. PMID 820877
- D. C. Chade u. a.: Immunomodulatory effects of recombinant BCG expressing pertussis toxin on TNF-alpha and IL-10 in a bladder cancer model. In: J Exp Clin Cancer Res 27, 2008, 78. PMID 19040745 (Open Access)
- M. D. Shelly u. a.: Intravesical bacillus Calmette-Guérin is superior to mitomycin C in reducing tumour recurrence in high-risk superficial bladder cancer: a meta-analysis of randomized trials. In: BJU Int 93, 2004, S. 485–490. PMID 15008714 (Review)
- M. D. Shelly u. a.: Intravesical bacillus Calmette-Guerin versus mitomycin C for Ta and T1 bladder cancer. In: Cochrane Database Syst Rev 3, 2003, CD003231. PMID 12917955 (Review)
- R. De Jager u. a.: Long-term complete remission in bladder carcinoma in situ with intravesical TICE bacillus Calmette Guerin. Overview analysis of six phase II clinical trials. In: Urology 38, 1991, S. 507–513. PMID 1836081
- F. M. Martin und A. M. Kamat: Definition and management of patients with bladder cancer who fail BCG therapy. In: Expert Rev Anticancer Ther 9, 2009, S. 815–820. PMID 19496718
- H. W. Herr und A. Morales: History of bacillus Calmette-Guerin and bladder cancer: an immunotherapy success story. In: The Journal of urology 179, 2008, S. 53–56. PMID 17997439 (Review)
- J. C. Kim und G. D. Steinberg: The limits of bacillus Calmette-Guerin for carcinoma in situ of the bladder. In: J Urol 165, 2001, S. 745–756. PMID 11176460 (Review)
- A. Böhle und S. Brandau: Immune mechanisms in bacillus Calmette-Guerin immunotherapy for superficial bladder cancer. In: J Urol 170, 2003, S. 964–969. PMID 12913751 (Review)
- C. L. Amling: Diagnosis and management of superficial bladder cancer. In: Curr Probl Cancer 25, 2001, S. 219–278. PMID 11514784 (Review)
- A. M. Jackson u. a.: Changes in urinary cytokines and soluble intercellular adhesion molecule–1 (ICAM-1) in bladder cancer patients after bacillus Calmette-Guerin (BCG) immunotherapy. In: Clin Exp Immunol 99, 1995, S. 369–375. PMID 7882559
- G. N. Thalmann u. a.: Urinary interleukin-8 and 18 predict the response of superficial bladder cancer to intravesical therapy with bacillus Calmette-Guerin. In: J Urol 164, 2000, S. 2129–2133. PMID 1106194
- S. Prescott u. a.: Intravesical Evans strain BCG therapy: quantitative immunohistochemical analysis of the immune response within the bladder wall. In: J Urol 147, 1991, S. 1636–1642. PMID 1593713
- A. Böhle u. a.: Effects of local bacillus Calmette-Guerin therapy in patients with bladder carcinoma on immunocompetent cells of the bladder wall. In: J Urol 144, 1990, S. 53–58. PMID 235918
- T. Higuchi u. a.: A possible mechanism of intravesical BCG therapy for human bladder carcinoma: involvement of innate effector cells for the inhibition of tumor growth. In: Cancer Immunol Immunother 58, 2009, s. 1245–1255. PMID 19139883 (Open Access)
- M. D. Shelly u. a.: Intravesical Bacillus Calmette-Guerin in Ta and T1 Bladder Cancer. In: Cochrane Database Syst Rev 4, 2000, CD001986. PMID 11034738 (Review)
- Science. 2006 Oct 6;314(5796):126-9. Epub 2006 Aug 31. Cancer regression in patients after transfer of genetically engineered lymphocytes. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA.
- N Engl J Med. 2014 Oct 16;371(16):1507-17. doi:10.1056/NEJMoa1407222. Chimeric antigen receptor T cells for sustained remissions in leukemia. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA.
- Cancer Immunol Immunother. 2014 Oct;63(10):1061-71. doi:10.1007/s00262-014-1575-2. Epub 2014 Jul 4. A phase I clinical trial combining dendritic cell vaccination with adoptive T cell transfer in patients with stage IV melanoma. Poschke I, Lövgren T, Adamson L, Nyström M, Andersson E, Hansson J, Tell R, Masucci GV, Kiessling R.
- Anticancer Res. 2013 Dec;33(12):5495-500. Wilms’ Tumor Gene 1 (WT1)--loaded dendritic cell immunotherapy in patients with uterine tumors: a phase I/II clinical trial. Coosemans A, Vanderstraeten A, Tuyaerts S, Verschuere T, Moerman P, Berneman ZN, Vergote I, Amant F, VAN Gool SW.
- Immunotherapy. 2012 Jul;4(7):703-18. doi:10.2217/imt.12.40. Dendritic cell engineering for tumor immunotherapy: from biology to clinical translation. Bhargava A, Mishra D, Banerjee S, Mishra PK.
- P. A. Bradbury und F. A. Shepherd: Immunotherapy for lung cancer. In: J Thorac Oncol 3, 2008, S. 164–170. PMID 18520304 (Review)
- B. Ebbell: The Papyrus Ebers: the greatest Egyptian medical document. Oxford University Press, 1937.
- S. A. Hoption Cann u. a.: Dr William Coley and tumour regression: a place in history or in the future. In: Postgraduate Medical Journal 79, 2003, S. 672–680. PMID 14707241 (Review)
- T. F. Murphy: Case studies in biomedical research ethics. MIT Press, 2004 ISBN 0-262-63286-1, S. 112.
- C. V. Ichim: Revisiting immunosurveillance and immunostimulation: Implications for cancer immunotherapy. In: J Transl Med 3, 2005, 8. PMID 15698481 (Review im Open Access)
- P. Bruns: Die Heilwirkung des Erysipels auf Geschwülste. In: Beitr Klin Chir 3, 1888, S. 443–466.
- P. Klärner: Regressionen des Yoshida-Sarkoms nach einmaliger Injektion eines Lipopolysaccharids aus E. Coli. In: Zeitschrift für Krebsforschung 62, 1958, S. 291–296.
- W. B. Coley: II. Contribution to the Knowledge of Sarcoma. In: Ann Surg 14, 1891, S. 199–220. PMID 17859590, PMC 1428624 (freier Volltext)
- D. B. Levine: The Hospital for the Ruptured and Crippled: William Bradley Coley, Third Surgeon-in-Chief 1925-1933. In: HSS J 4, 2008, S. 1–9. doi:10.1007/s11420-007-9063-2 PMID 18751855 PMC 2504278 (freier Volltext)
- D. B. Levine: The Hospital for the Ruptured and Crippled: Knight to Gibney. 1870–1887. In: HSS J 2, 2006, S. 1–6 .
- W. B. Coley: Contribution to the knowledge of sarcoma. In: Ann Surg 14, 1891, S. 199–220.
- W. B. Coley: The Treatment of Malignant Tumors by Repeated Innoculations of Erysipelas: With a Report of Ten Original Cases. In: American Journal of the Medical Sciences 10, 1893, S. 487–511.
- B. Wiemann und C. O. Starnes: Coley’s toxins, tumor necrosis factor and cancer research: a historical perspective. In: Pharmacol Ther 64, 1994, S. 529–564. PMID 7724661
- W. B. Coley: End results in Hodgkin’s disease and lymphosarcom treated by the mixed toxins of erysipelas nad bacillus prodigiosus, alon or combined with radiation. In: Ann Surg 88, 1928, S. 641–667. PMID 17865976
- E. F. McCarthy: The Toxins of William B. Coley and the Treatment of Bone and Soft-Tissue Sarcomas. In: Iowa Orthop J 26, 2006, S. 154–158. PMID 16789469 PMC 1888599 (freier Volltext)
- J. Bickels u. a.: Coley’s toxin: historical perspective. (Memento des Originals vom 1. Februar 2014 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. In: Isr Med Assc J 4, 2002, S. 471–472. PMID 12073431
- P. N. Leech: Erysipelas and Prodigiosus toxins (Coley). In: JAMA 103, 1934, S. 1067–1069.
- A. Pollack: A Revival for Immunity. In: New York Times Ausgabe vom 5. Oktober 2005
- P. Ehrlich: Über den jetzigen Stand der Karzinomforschung. In: Ned Tijdschr Geneeskd 5, 1909, S. 273–290.
- H. F. von Doerr u. a.: Pathologie des Thymus. Verlag Springer, 1998, ISBN 3-540-64065-7. S. 103.
- R. Pearl: Cancer and tuberculosis. In: Am J Hygiene 9, 1929, S. 97
- L. Thomas: Cellular and Humoral Aspects of Hypersensitivity. H. S. Lawrence (Editor), Hoeber-Harper, 1959.
- F. M. Burnet: The concept of immunological surveillance. In: Prog Exp Tumor Res 13, 1970, S. 1–27. PMID 4921480
- E. Coeugniet: Editorial. In: Onkologie Juni 1989, S. 3–4.
- S. Höpner: Charakterisierung einer hABL-spezifischen CD4+-T-Zellantwort und die Anwendung des AdEtOH als Katalysator der Peptidbeladung. Dissertation, FU Berlin, 2008
- J. Rygaard und C. O. Povlsen: The mouse mutant nude does not develop spontaneous tumours. An argument against immunological surveillance. In: Acta Pathol Microbiol Scand [B] Microbiol Immunol 82, 1974, S. 99–106. PMID 4597815
- O. Stutman: Tumor development after 3-methylcholanthrene in immunologically deficient athymic-nude mice. In: Science 183, 1974, S. 534–536. PMID 4588620
- J. R. Maleckar L. A. Sherman: The composition of the T cell receptor repertoire in nude mice. In: J Immunol 138, 1987, S. 3873–3876. PMID 2953792
- S. Ikehara u. a.: Functional T cells in athymic nude mice. In: PNAS 81, 1984, S. 886–888. PMID 6608104
- R. B. Herberman und H. T. Holden: Natural cell-mediated immunity. In: Adv Cancer Res 27, 1978, S. 305–377. PMID 356546 (Review)
- C. Huber (Herausgeber) u. a.: Krebsimmuntherapien. Deutscher Ärzteverlag, 2007, ISBN 3-7691-1212-1, S. 5.
- C. Y. Li u. a.: Cytokine and immuno-gene therapy for solid tumors. (PDF; 746 kB) In: Cell Mol Immunol 2, 2005, S. 81–91. PMID 16191413 (Review)
- James P. Allison und Carl H. June erhalten Paul Ehrlich- und Ludwig Darmstaedter-Preis 2015, Pressemeldung der Paul Ehrlich-Stiftung vom 29. Januar 2015, abgerufen am 6. Februar 2015