Drug Targeting
Drug Targeting oder Targeted Drug Delivery, manchmal auch Smart Drug Delivery[1] genannt, ist die zielgerichtete und selektive Anreicherung oder Freisetzung eines Arzneistoffs an einem oder mehreren gewünschten Wirkorten. Auf diese Weise sollen die Effektivität der Wirkung erhöht und gleichzeitig systemische Nebenwirkungen verringert werden. Ein Targeting eines Arzneistoffs ist mit Hilfe chemischer Modifikationen des Wirkstoffes, mit Hilfe der Biotechnologie oder mit Hilfe der pharmazeutischen Technologie möglich.
Zweck
Für eine gezielte Versorgung des zu behandelnden Gewebes gibt es pharmazeutische, biopharmazeutische, pharmakodynamische, pharmakokinetische und pharmakoökonomische Gründe. Beispiele hierfür sind eine verminderte Anzahl unerwünschter Arzneimittelwirkungen, eine veränderte und gezieltere Wirkstofffreisetzung, weniger benötigte Dosen oder eine erhöhte Patientencompliance.[1] Zusätzlich kann eine ausreichende Versorgung der betroffenen Zellen, des betroffenen Gewebes oder der betroffenen Organe unter Verwendung klassischer Methoden bei zahlreichen Erkrankungen, wie beispielsweise zentralnervöse Störungen, rheumatoide Arthritis, Tumorerkrankungen und Tuberkulose, erschwert sein. Ein Drug Targeting kann in diesen Fällen ebenfalls hilfreich sein.
Strategien beim Drug Targeting
Im Allgemeinen können aktives und passives Drug Targeting unterschieden werden.[1]
Passive Targeting
Das Prinzip beim Passive Targeting basiert auf der Akkumulation des Wirkstoffes am gewünschten Ort oder Gewebe. Bei Tumoren kann beispielsweise die Induktion von Angiogenese, die erhöhte Aktivität proliferativen Signallings oder das Deregulieren des zellulären Energiestoffwechsels (s. Warburg-Effekt) der Tumorzellen ausgenutzt werden.[2]
Active Targeting
Beim Active Targeting werden Ligand-Rezeptor-Wechselwirkungen beeinflusst. Diese können nämlich nur bei Abständen von kleiner als ungefähr 0,5 mm entstehen, was die Drug Delivery Systeme hervorrufen.
Methoden
Targeting aufgrund physikochemischer Eigenschaften des Arzneistoffs
Die einfachste Form des Drug Targetings besteht in der Optimierung der physikochemischen Eigenschaften, insbesondere der Löslichkeit, der Lipophilie und der Azidität oder Basizität des Arzneistoffs. Ein solches Drug Targeting ist im Allgemeinen nicht von einem Arzneistoffträger abhängig. Die Selektivität für ein bestimmtes Zielgewebe ist hingegen in der Regel beschränkt.
Ein solcher, auf den physikochemischen Stoffeigenschaften beruhender Targeting-Effekt wird für die sauren Nichtopioid-Analgetika diskutiert. Saure Nichtopioid-Analgetika, wie Acetylsalicylsäure, Ibuprofen, Naproxen, Diclofenac, Indometacin und weiteren Vertreter, liegen bei physiologischem pH-Wert überwiegend in ihrer deprotonierten anionischen Form vor. Im sauren, entzündeten Gewebe hingegen können sie sich anreichern, da sie hier in relevanter Menge protoniert und somit immobilisiert vorliegen. Diese Eigenschaft saurer Nichtopioid-Analgetika wird als Begründung für ihre therapeutische Überlegenheit gegenüber nichtsauren Analgetika, wie Phenazon, Metamizol und Paracetamol, bei der Behandlung entzündlicher Erkrankungen angeführt.[3]
Target-selektive Aktivierung von Prodrugs
Eine weitere Option sind inaktive Arzneistoffvorstufen (Prodrugs), die selektiv innerhalb des Zielgewebes in ihre aktive Form (aktive Metabolite) umgewandelt werden. Ein Beispiel hierfür sind die Protonenpumpenhemmer, zu denen beispielsweise Omeprazol zählt.[4] Protonenpumpenhemmer sind Prodrugs, die im stark sauren Milieu, insbesondere an der Oberfläche der Magensäure produzierenden Parietalzellen, aktiviert werden und als Folge Proteine, wie die H+/K+-ATPase (Protonenpumpe), durch kovalente Bindung inaktivieren.
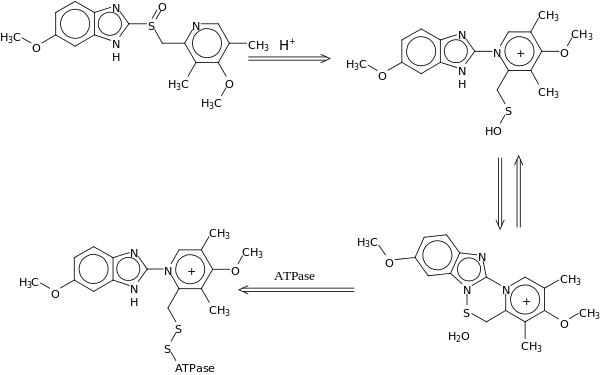
Ein weiteres Beispiel für ein Drug Targeting mit Hilfe von Prodrugs, die selektiv im Zielgewebe aktiviert werden, sind die Antibiotika aus der Gruppe der Nitroimidazole, wie beispielsweise Metronidazol. Nitroimidazole sind insbesondere unter anaeroben Bedingungen wirksam und haben ein breites Wirkspektrum gegen anaerobe Keime. Im anaeroben Milieu wird das Prodrug Metronidazol von den Bakterien enzymatisch unter Beteiligung von Ferredoxin in sein hochreaktives Zwischenprodukt N-(2-Hydroxyethyl)-oxamidsäure gespalten. Dieses Zwischenprodukt führt zu DNA-Strangbrüche innerhalb der bakteriellen DNA und ist somit für die bakterizide Wirkung von Metronidazol verantwortlich.
Vektorisierung
Eine Form des Drug Targetings ist die Vektorisierung, die Konjugation des zu verabreichenden Arzneistoffs an ein Molekül, von dem bekannt ist, dass es an die Zielzellen bindet. Zu diesem Zweck können Arzneistoffe an Antikörper, Transferrin oder andere Biomoleküle gekoppelt werden. Alternativ dazu können auch synthetische Polymere eingesetzt werden.
Antikörperkonjugate
Antikörperkonjugate sind makromolekulare Arzneistoffe, bei denen mindestens ein Molekül des eigentlichen Wirkstoffs über eine kovalente Bindung an einen Antikörper gebunden ist. Der Antikörper ist üblicherweise gegen ein Oberflächenmolekül gerichtet, das für die Zielzellen oder das Zielgewebe spezifisch ist. Nach Anbindung an die Zielzellen können die Konjugate optional über eine rezeptorvermittelte Endozytose über Vesikel in die Zielzellen aufgenommen werden. Arzneistoffe, die an Antikörper gekoppelt sind, werden insbesondere in der Chemotherapie maligner Tumoren eingesetzt. Beispiele hierfür sind Gemtuzumab-Ozogamicin und Ibritumomab-Tiuxetan.
Im Falle des Gemtuzumab-Ozogamicins wurde der monoklonale, gegen das CD33-Antigen gerichtete Antikörper Gemtuzumab an das Zytostatikum Ozogamicin, einem Verwandten des Calicheamicins gekoppelt. Für eine Wirksamkeit ist eine Aufnahme des Antikörper-Zytostatikum-Konjugats in die Zelle Voraussetzung.[5] Durch die Konjugation und das Targeting wird die systemische Toxizität von Ozogamicin reduziert.
Ibritumomab-Tiuxetan wird in der Radioimmuntherapie verschiedener maligner B-Zell-Lymphome (Lymphdrüsenkrebs) angewendet. Es ist ein Konjugat aus einem monoklonalen Antikörper, der gegen das CD20-Antigen an der Oberfläche von B-Lymphozyten gerichtet ist, und dem Chelator Tiuxetan (ein DTPA-Derivat), der beispielsweise das radioaktive Isotop Yttrium-90 komplexieren kann. Dieses Targeting radioaktiver Isotope entspricht zugleich einer zielgerichteten Strahlentherapie.
Neuere Entwicklungen schließen Konjugate bestehend aus einem Wirkstoff und Antikörperfragmenten, wie beispielsweise F(ab)2-Fragmente, Fab-Fragmente und Einzeldomänenantikörper oder Antikörpermimetika, wie z. B. Anticaline, ein. Diese Konjugate sollen sich durch eine geringere Immunogenität und eine verbesserte Gewebepermeabilität auszeichnen.[6]
Peptidkonjugate
Auch die Konjugation eines Arzneistoffs an ein Peptid oder an niedermolekulare Substanzen kann zu einer Anreicherung des Wirkstoffs im Zielgewebe beitragen und optional eine Einschleusung in die Zielzellen ermöglichen. Als Vektoren für Wirkstoffe, wie beispielsweise Doxorubicin, sind insbesondere die sogenannten zellpenetrierenden Peptide für die Forschung und Entwicklung von Interesse. Für ihre endozytoische Aufnahme spielen je nach pharmakologischen und physikochemischen Eigenschaften des Peptids neben rezeptorvermittelten auch unspezifische, adsorptionsvermittelte Mechanismen eine Rolle. Letztere können insbesondere bei basischen Peptiden beobachtet werden und führen über elektrostatische Wechselwirkungen zwischen der von Glykoproteinen negativ geladenen Zelloberfläche und positiv geladenen Vektorpeptiden zur unspezifischen Bindung an die Zelloberfläche, in deren Folge eine vesikuläre Aufnahme in das Zytoplasma erfolgt.
Experimentell finden unter anderem basische Protegrin-Abkömmlinge, wie beispielsweise Syn-B,[7][8] und das aus der Homöodomäne von Antennapedia, einem Transkriptionsfaktor von Drosophila, abgeleitete Penetratin[9] für ein ZNS-Targeting Anwendung. Ein anderer Peptid-Vektor ist das aus elf überwiegend basischen Aminosäuren bestehende und aus der Transduktionsdomäne des HI-Virus isolierte HIV-TAT (engl. Trans-Activator of Transcription).[10][11] Ein Peptid mit ähnlichen Eigenschaften ist das aus 27 Aminosäuren aufgebaute Transportan.[12]
Polymerkonjugate
Eine weitere Möglichkeit besteht in der Konjugation eines Arzneistoffs mit einem löslichen Polymer, wie beispielsweise Cyclodextrin, Polyglutamat, Polyaspartat, Hydroxypropylmethacrylamid (HPMA) oder Polyethylenglycol (PEG).[13] Optimalerweise ist der Arzneistoff über einen hydrolysierbaren Linker mit dem Polymer verbunden und in seiner gebundenen Form pharmakologisch inaktiv (Prodrug). Nach Aufnahme in die Zielzellen durch Endozytose und unter Einwirkung lysosomaler Enzyme kann der Arzneistoff aus dem Konjugat in seiner aktiven Form freigesetzt werden. Auf diese Weise lässt sich zusätzlich eine verzögerte Wirkstofffreisetzung erzielen. Die Targetselektivität der Polymerkonjugate ist jedoch meist limitiert, da sie von den physikochemischen Eigenschaften des Polymers geprägt wird. Ungeachtet dessen zeigen viele Polymerkonjugate auf Grund des EPR-Effektes eine Tendenz im Tumorgewebe zu akkumulieren.[14] Die Gewebeselektivität von Arzneistoff-Polymer-Konjugaten kann durch eine weitere Konjugation mit Antikörpern gesteigert werden.[15]
Mit dem Ziel eines Drug Targetings wurden zahlreiche potenzielle Arzneistoffe entwickelt, die den EPR-Effekt ausnutzen. Polymerkonjugate, wie beispielsweise HPMA-Doxorubicin, HPMA-Camptothecin, HPMA-Paclitaxel und Pegamotecan (PEG-Camptothecin) befinden sich derzeit in der klinischen Erprobung.[13] Für Paclitaxel-Poliglumex, ein Konjugat aus Paclitaxel und Polyglutamat, wird die arzneimittelrechtliche Zulassung demnächst erwartet.[16]
Partikuläre Träger
Partikuläre Träger stellen eine Möglichkeit der pharmazeutischen Technologie dar, einen Arzneistoff zielgerichtet zu transportieren. Für ein Drug Targeting kann wie im Falle der Polymerkonjugate der EPR-Effekt ausgenutzt werden, um ein Arzneistoff in das Tumorgewebe zu transportieren. Zusätzlich können partikuläre Träger mit zell- oder gewebespezifischen Antikörpern oder zellpenetrierenden Peptiden[17] konjugiert werden, um den Arzneistoff selektiv in das Zielgewebe zu transportieren. Der Arzneistoff ist in der Regel physikalisch im oder an den Träger gebunden und kann nach Erreichung seines Bestimmungsorts aus seinem Träger freigesetzt werden. Im Inneren des Trägers ist der Arzneistoff zusätzlich vor einer Metabolisierung geschützt. Zu den am häufigsten für ein Targeting eingesetzten Trägern gehören Mizellen, Nanopartikel und Liposomen.
Liposomen
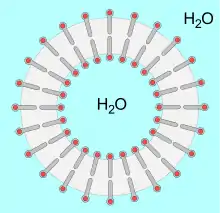
Liposomen sind partikuläre Träger von einer Größe von 50 bis 1000 nm, deren wässrige innere Phase durch eine Phospholipid-Doppelschicht von der äußeren Phase abgetrennt ist. Im Inneren eines Liposoms kann ein wasserlöslicher Arzneistoff verkapselt werden. Alternativ können fettlösliche Arzneistoffe in begrenzter Menge in der Phospolipid-Membran angereichert werden. Künstliche Liposomen sind weitgehend stabil im Organismus und besitzen wie ihre natürlich vorkommenden Vorbilder eine geringe Toxizität und Allergenität. Ihr Abbau erfolgt bevorzugt nach endozytotischer Aufnahme in den Zellen des Reticuloendothelialen Systems. Eine Modifizierung der Liposomenoberfläche mit Polyethylenglykol, die sogenannte PEGylierung, kann Liposomen maskieren und so vor einem Abbau schützen („Stealth Liposomen“).[18] Optional kann ein zielsuchender Ligand, beispielsweise ein Antikörper, in der Liposomen-Hülle verankert werden.[19][20][21]
Einige Beispiele für liposomale Arzneimittel, die bereits zur Therapie zugelassen wurden, sind in der nachstehenden Tabelle aufgeführt. Weitere liposomale Arzneimittel, wie beispielsweise liposomal verkapseltes Cisplatin, Lurtotecan, Tretinoin und Vincristin befinden sich derzeit in der klinischen Erprobung. Liposomen finden darüber hinaus bei der Transfektion, Einschleusung von DNA in Zellen, Anwendung und stellen somit potenzielle Vehikel in der Gentherapie dar. Für diese Anwendung, die auch als Lipofektion bezeichnet wird, werden insbesondere Liposomen eingesetzt, die aus kationischen Lipiden aufgebaut sind.
| Arzneimittel | Arzneistoff | Zielgewebe | Indikation |
|---|---|---|---|
| AmBisome | Amphotericin B | schwere systemische oder tiefe Mykosen[22] | |
| DaunoXome | Daunorubicin | Tumorgewebe | Kaposi-Sarkom |
| Myocet, Doxil | Doxorubicin | Tumorgewebe | Mammakarzinom[23] |
| Caelyx | Doxorubicin | Tumorgewebe | Kaposi-Sarkom, Ovarialkarzinom, Mammakarzinom, multiples Myelom[24] |
Mizellen
Mizellen stellen eine Möglichkeit dar, partikuläre Träger mithilfe von amphiphilen Blockcopolymeren (Polymeren mit verschiedenen Arten von Monomeren) in wässriger Lösung herzustellen.[25] Dabei können der Wirkstoff oder die Wirkstoffe sich innerhalb der Blockcopolymere (5–50 nm) befinden und von diesen zu Orten transportiert werden, an denen sie sich in der Regel nicht lösen würden. Durch Bildung einer Hülle um den Wirkstoff herum kommt es außerdem zum Schutz vor Hydrolyse oder enzymatischem Abbau.
Nanopartikel
Nanopartikel sind Carrier, die aus natürlichen oder synthetischen Polymeren mit einer Größe von ungefähr 10 bis 1000 nm bestehen. Der Wirkstoff kann dabei entweder als Kristall im Nanopartikel angelagert (s. Mischkristall), an dessen Oberfläche adsorbiert oder chemisch gebunden sein.
Ein potenzieller Vorteil dieser Drug-Delivery-Systemen ist, dass sie unter bestimmten Bedingungen die Blut-Hirn-Schranke überwinden können. Das Hexapeptid Dalargin (Tyr-D-Ala-Gly-Phe-Leu-Arg) war der erste Wirkstoff, der mithilfe von Polysorbat-80-beschichteten Polybutylcyanoacrylat-Nanopartikeln die Blut-Hirn-Schranke in vivo überwinden konnte.[26] Es handelt sich dabei um ein Leu-Enkephalin-Analogon, welches eine Aktivität an Opioid-Rezeptoren aufweist und folglich die Schmerzempfindung beeinflussen kann. Der Mechanismus beim Überschreiten der Blut-Hirn-Schranke ist noch nicht genau verstanden. Jedoch wird vermutet, dass es Zusammenhänge mit der Partikelgröße und der Struktur gibt. So werden beispielsweise PEG-überzogene Nanopartikel, welche die Struktur von LDL (Low-Density-Lipoprotein) nachahmen, in das Gehirn transportiert.
Einzelnachweise
- Nidhi Mishra, Prerna Pant, Ankit Porwal, Juhi Jaiswal, Mohd Aquib Samad, Suraj Tiwari: Targeted Drug Delivery: A Review. In: American Journal Of PharmTech Research. 2016
- D. Hanahan, R. A. Weinberg: Hallmarks of cancer: the next generation. In: Cell. 144, 2011, S. 646–674.
- K. Brune, K. D. Rainsford, A. Schweitzer: Biodistribution of mild analgesics. In: Br J Clin Pharmacol. 10 Suppl 2, Oktober 1980, S. 279S–284S, PMID 6969084, PMC 1430188 (freier Volltext).
- Per Lindberg, Endar Carlsson: Esomeprazole in the framework of proton-pump inhibitor development. In: János Fischer, Robin Ganellin (Hrsg.): Analogue-based drug discovery. Band 1. John Wiley & Sons, 2006, ISBN 3-527-31257-9, S. 81–114.
- V. H. van Der Velden, J. G. te Marvelde, P. G. Hoogeveen u. a.: Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: in vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. In: Blood. Band 97, Nr. 10, Mai 2001, S. 3197–3204, PMID 11342449.
- P. Holliger, P. J. Hudson: Engineered antibody fragments and the rise of single domains. In: Nat. Biotechnol. Band 23, Nr. 9, September 2005, S. 1126–36, doi:10.1038/nbt1142, PMID 16151406.
- C. Rousselle, P. Clair, J. M. Lefauconnier, M. Kaczorek, J. M. Scherrmann, J. Temsamani: New advances in the transport of doxorubicin through the blood-brain barrier by a peptide vector-mediated strategy. In: Mol. Pharmacol. Band 57, Nr. 4, April 2000, S. 679–686, PMID 10727512.
- C. Rousselle, M. Smirnova, P. Clair u. a.: Enhanced delivery of doxorubicin into the brain via a peptide-vector-mediated strategy: saturation kinetics and specificity. In: J. Pharmacol. Exp. Ther. Band 296, Nr. 1, Januar 2001, S. 124–131, PMID 11123372.
- B. Christiaens, S. Symoens, S. Verheyden u. a.: Tryptophan fluorescence study of the interaction of penetratin peptides with model membranes. In: Eur. J. Biochem. Band 269, Nr. 12, Juni 2002, S. 2918–26, doi:10.1046/j.1432-1033.2002.02963.x, PMID 12071955.
- S. R. Schwarze, A. Ho, A. Vocero-Akbani, S. F. Dowdy: In vivo protein transduction: delivery of a biologically active protein into the mouse. In: Science. Band 285, Nr. 5433, September 1999, S. 1569–1572, PMID 10477521.
- J. M. Scherrmann: Drug delivery to brain via the blood-brain barrier. In: Vascul. Pharmacol. Band 38, Nr. 6, Juni 2002, S. 349–354, doi:10.1016/S1537-1891(02)00202-1, PMID 12529929.
- M. Pooga, M. Hällbrink, M. Zorko, U. Langel: Cell penetration by transportan. In: FASEB J. Band 12, Nr. 1, Januar 1998, S. 67–77, PMID 9438412.
- C. Li, S. Wallace: Polymer-drug conjugates: recent development in clinical oncology. In: Adv Drug Deliv Rev. Band 60, Nr. 8, Mai 2008, S. 886–98, doi:10.1016/j.addr.2007.11.009, PMID 18374448, PMC 2432086 (freier Volltext).
- M. J. Vicent, R. Duncan: Polymer conjugates: nanosized medicines for treating cancer. In: Trends Biotechnol. Band 24, Nr. 1, Januar 2006, S. 39–47, doi:10.1016/j.tibtech.2005.11.006, PMID 16307811.
- Z. R. Lu, P. Kopecková, J. Kopecek: Polymerizable Fab' antibody fragments for targeting of anticancer drugs. In: Nat. Biotechnol. Band 17, Nr. 11, November 1999, S. 1101–1104, doi:10.1038/15085, PMID 10545917.
- CTI - Herstellerinformationen zu Opaxio (Memento vom 3. Dezember 2008 im Internet Archive)
- V. P. Torchilin: Cell penetrating peptide-modified pharmaceutical nanocarriers for intracellular drug and gene delivery. In: Biopolymers. Band 90, Nr. 5, 2008, S. 604–610, doi:10.1002/bip.20989, PMID 18381624.
- M. L. Immordino, F. Dosio, L. Cattel: Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. In: Int J Nanomedicine. Band 1, Nr. 3, 2006, S. 297–315, PMID 17717971, PMC 2426795 (freier Volltext).
- Vladimir Torchilin, Volkmar Weissig: In: Liposomes: A Practical Approach. 2. Auflage. Oxford University Press, 2003, ISBN 0-19-963654-0.
- M. Thöle: Arzneistofftransport an der Blut-Hirn-Schranke: Drug Targeting mit liposomalen Konjugaten. Dissertation. 2000.
- A. Sharma, E. Mayhew, L. Bolcsak, C. Cavanaugh, P. Harmon, A. Janoff, R. J. Bernacki: Activity of paclitaxel liposome formulations against human ovarian tumor xenografts. In: Int J Cancer. 71, 1997, S. 103–107. PMID 9096672.
- Fachinformation AmBisome® 50 mg Pulver zur Herstellung einer Infusionslösung. Gilead. Stand April 2009.
- Fachinformation Myocet®. Cephalon. Stand November 2008.
- Fachinformation Caelyx® 2 mg/ml. Stand Dezember 2007.
- G. Tiwari, R. Tiwari, B. Sriwastawa u. a.: Drug delivery systems: An updated review. In: International Journal of Pharmaceutical Investigation. 2(1), 2012, S. 2–11.
- Jörg Kreuter: Nanoparticulate Carriers for Drug Delivery to the Brain. In: V. P. Torchilin (Hrsg.): Nanoparticulates as Drug Carriers. World Scientific, 2006, ISBN 1-86094-630-5, S. 529 (eingeschränkte Vorschau in der Google-Buchsuche).