Komplementsystem
Das Komplementsystem ist ein System von Plasmaproteinen, das im Zuge der Immunantwort auf zahlreichen Oberflächen von Mikroorganismen aktiviert werden kann. Ursprünglich wurde es als ergänzender (komplementärer) Teil der Antikörperantwort entdeckt, doch ist heute bekannt, dass es auch am angeborenen Immunsystem beteiligt ist.
Die mehr als 30 Proteine des menschlichen Komplementsystems sind im Blutplasma gelöst oder zellgebunden und dienen der Abwehr von Mikroorganismen (z. B. Bakterien, Pilze, Parasiten). Sie haben jedoch auch stark zellzerstörende Eigenschaften und können, wenn sie unreguliert wirken, im Verlauf vieler Krankheiten (z. B. Glomerulonephritis, hämolytisch-urämisches Syndrom, Herzinfarkt, systemischer Lupus erythematodes, Rheumatoide Arthritis) für Gewebsschäden verantwortlich sein.
Begriff
Der Ausdruck „Komplement“ wurde um 1890 von Paul Ehrlich eingeführt. Seiner Theorie zufolge bestand das Immunsystem aus Zellen mit spezifischen Rezeptoren, die Antigene erkennen können. Nach Antigenkontakt werden diese Rezeptoren formiert und zur Verteilung ins Blut abgegeben. Diese heutzutage als Antikörper bezeichneten Rezeptoren nannte Ehrlich Ambozeptoren. Deren Funktion ist es, sowohl das Antigen als auch eine hitze-empfindliche Komponente des Blutserums zu erkennen (die Ehrlich als Komplement bezeichnete), da sie die Funktion der zellulären Immunabwehr ergänzen. Auch wenn Ehrlich den Begriff Komplement prägte, geht die eigentliche Beschreibung des Komplement auf Jules Bordet zurück: Er entdeckte, dass das Komplement sowohl in Zusammenhang mit Antikörpern als auch allein wirken kann.
Wirkungsweise
Die Hauptaufgabe des Komplementsystems besteht darin, die Oberfläche von Krankheitserregern zu bedecken, um so den Phagozyten auch die Zerstörung jener Krankheitserreger zu ermöglichen, die sie sonst nicht erkennen würden (Opsonisierung). Daneben löst es eine Reihe von Entzündungsreaktionen aus, die den Kampf gegen die Infektion unterstützen. Die Fragmente einiger Komplementproteine wirken als Chemokine, die weitere Phagozyten zum Infektionsherd locken. Eine weitere Funktion ist die direkte Zerstörung von Bakterien durch das Einfügen von Poren in deren Zellmembranen.
Ein großer Teil der Komplementproteine sind sogenannte Zymogene. Das sind Proteine, in diesem Fall Proteasen, die ihrerseits durch limitierte Proteolyse aktiviert werden. Diese Zymogene kommen normalerweise überall im Körper vor, ohne dass es zu einer Reaktion kommt. Im Falle einer Infektion werden sie jedoch lokal aktiviert und aktivieren weitere Zymogene durch deren Spaltung. Dadurch wird eine Kaskade von Zymogenaktivierungen ausgelöst, wobei aus wenigen früh aktivierten Molekülen viele später aktivierte werden, was zu einer Verstärkung der Antwort führt.
Bestandteile des Komplementsystems
Direkt an den Signalwegen des Komplementsystems beteiligt sind folgende Proteine: die Komplementfaktoren C1 bis C9, das Mannose-bindende Lektin (MBL) und die an C1 bzw. MBL gebundenen Serinproteasen C1r und C1s bzw. MASP-1 bis 3 (engl. MBL-associated serine proteases). Durch Protease-vermittelte Spaltung der Komplementfaktoren C1 bis C5 und Zusammenlagerungen mit den Faktoren C6 bis C9 entsteht eine Vielzahl an Proteinen und Proteinkomplexen. Zu diesen gehören beispielsweise die Anaphylatoxine C3a, C5a und C4a mit gefäßerweiternder, bronchokonstriktorischer und chemotaktischer Wirkung (Entzündungsreaktion) und der Membranangriffskomplex (engl. Membrane Attack Complex (MAC)). Negativregulatoren des Systems sind der C1-Inhibitor, Faktor H, Faktor I, C4bp, CD35, CD46, CD55, CD59 und Vitronektin. Als Aktivatoren wirken Properdin und Cobra Venom Factor.
Ablauf und Wirkung der Komplement-Aktivierung
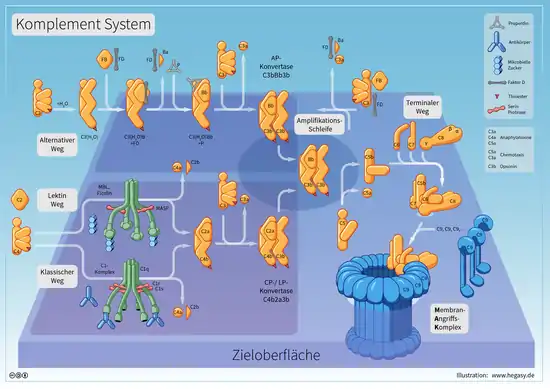
Man unterscheidet drei Wege, durch die das Komplementsystem aktiviert wird:
- Den oft über Antikörper vermittelten klassischen Weg.
- Den über Mannose-bindendes Lektin aktivierten Lektin-Weg.
- Den spontanen und Antikörper-unabhängigen alternativen Weg.
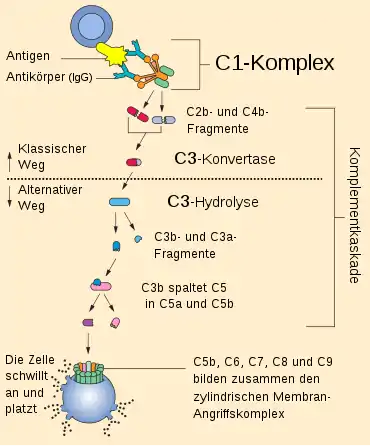
Das Produkt aller drei Wege ist eine als C3-Konvertase bezeichnete Serinprotease auf der Oberfläche der Zielzelle. Die von ihr ausgelöste Spaltungskaskade führt zu chemotaktischer Anlockung von Leukozyten (insbesondere Makrophagen), verstärkter Phagozytose, und letztendlich zur Lyse der Zielzelle. Spaltprodukte der Komplementfaktoren C1 bis C5, die in den einzelnen Wegen entstehen, wirken zusätzlich als Anaphylatoxine und vermitteln eine Entzündungsreaktion.
Der klassische Weg
Am klassischen Aktivierungsweg des Komplementsystems sind neun Glykoproteine (C1-C9) beteiligt. Diese haben Molekülmassen von 24 bis 410 kDa und werden nach Bildung in der Leber, in kleinem Umfang auch in Lymphocyten, Makrophagen und Fibroblasten, in die Blutbahn sezerniert, wo sie etwa 10 % der Globulinfraktion ausmachen. Der Komplementfaktor C1 ist das erste Komplementprotein des klassischen Weges und besteht aus dem sechsköpfigen Kollektin C1q und zwei Molekülen C1s und zwei Molekülen C1r (Abb. unter[1]). C1q besitzt mehrere Bindungsdomänen für Antigen gebundene Antikörper (IgG und IgM). Für die Aktivierung der an C1q gebundenen Serin-Proteasen (C1r und C1s) sind zwei 40 nm voneinander entfernte Ig-Fc-Regionen nötig. Deswegen reicht bei IgM-Antikörpern ein Molekül aus, bei IgG-Antikörpern sind dagegen mehrere, idealerweise sechs, Moleküle erforderlich;[2] IgA-, IgE- oder IgD-Antikörper können den klassischen Weg nicht aktivieren. Freie Antikörper führen daher nicht zur Aktivierung. C1q kann allerdings auch direkt an die Oberfläche von Krankheitserregern binden und den klassischen Weg so auch ohne die Hilfe von Antikörpern einleiten.
Die klassische Aktivierung kann auch durch DNA, Kollagen und CRP (C-reaktives Protein) ausgelöst werden.
Nach ihrer Aktivierung katalysiert die Serinprotease C1s dann die beiden Startreaktionen des klassischen Weges. Eine Spaltung von C2 in C2a und C2b und eine weitere von C4 in C4a und C4b. C2a und C4b lagern sich zum C4b2a-Komplex zusammen und bilden so die „C3-Konvertase des klassischen Weges“. C4b2a3b bildet die C5-Konvertase, die C5 in C5a und C5b spaltet. C3a und C5a diffundieren und wirken wiederum als Anaphylatoxin.
Bei der klassischen Aktivierung des Komplementsystems wird neben C4a und C4b in weiterer Folge auch C4d abgespalten, welches kovalent an das Endothel, an dem die Komplementreaktion stattgefunden hat, binden kann. Die Funktion des C4d ist derzeit noch nicht verstanden, es dient aber in der Biopsie als Marker und diagnostisches Mittel der Antikörper induzierten Transplantatabstoßung.
Die Aktivierung von C1 wird durch ein Plasmaprotein, den C1-Inhibitor, kontrolliert, der an den aktiven Enzymteil von C1 (C1r/s) bindet und ihn dadurch von C1q abtrennt. C4b-binding protein inhibiert die Bildung der C3 Konvertase (C4b2a-Komplex) über den klassischen und Lektin-Weg, indem es als Co-Faktor für Faktor I wirkt.[3]
Der Lektin-Weg
Im Lektin-Weg bindet das Mannose-bindende Lektin (MBL) an Mannose bzw. Fikolin an N-Acetylglucosamin auf der pathogenen Oberfläche (z. B. bakterielles Peptidoglykan) und aktiviert die MBL-assoziierten Serinproteasen MASP-1, MASP-2 und MASP-3. Diese katalysieren dieselben Reaktionen wie im klassischen Weg. Auch hier bilden wieder C4b und C2a ein C4b2a-Heterodimer und damit ebenfalls die „C3-Konvertase des klassischen Weges“.
| Schema des alternativen Weges |
|---|

Der alternative Weg
Der alternative Weg führt zur Bildung der „C3-Konvertase des alternativen Weges“. Ausgelöst wird dieser Weg durch den spontanen Zerfall des instabilen Komplementfaktors C3 in C3a und C3b. C3a diffundiert und besitzt eine chemotaktische und entzündungsauslösende Wirkung als Anaphylatoxin. C3b bindet kovalent an eine Zelloberfläche. Freies C3b wird durch Faktor H und Faktor I inaktiviert. Bindet C3b an körpereigene Zellen, wird es ebenfalls relativ rasch durch Regulatorproteine inaktiviert oder abgebaut. Auf pathogenen Oberflächen bleibt es dagegen aktiv und bindet Faktor B, der von Faktor D (Plasmaprotease) in Ba und Bb gespalten wird. Der entstehende Komplex C3bBb wird als „C3-Konvertase des alternativen Weges“ bezeichnet. Er ist sehr instabil und zerfällt, wenn er nicht von Properdin stabilisiert wird.
C3-Konvertase ausgelöste Reaktionen
Die im alternativen, klassischen und Lektin-Weg gebildeten C3-Konvertasen, C3bBb und C4b2a, spalten nun mit hoher Aktivität C3 in C3b und C3a. Die entstehenden C3b-Moleküle haben nun im Wesentlichen drei Möglichkeiten:
- Sie finden keine geeignete Oberfläche an die sie binden können und werden inaktiviert durch CD46.
- Die Moleküle lagern sich an die Zelloberfläche einer Zielzelle an und führen so zu einem weiteren „Start“ des alternativen Weges. Eine positive Rückkopplung entsteht. Außerdem wirken sie als Opsonine und markieren die Zielzelle als lohnendes Ziel zur Phagozytose.
- Einige der Moleküle binden an eine C3-Konvertase (C4b2a bzw. C3bBb). Die hierbei entstehenden trimolekularen Komplexe C4b2a3b und "C3bBb3b + Properdin" spalten nun nicht mehr C3, sondern C5. Daher werden sie jetzt als „C5-Konvertasen des klassischen bzw. alternativen Weges“ bezeichnet.
Die beiden Produkte der C5-Spaltung fungieren einerseits als Anaphylatoxin und chemotaktischer Lockstoff (C5a) und andererseits leiten sie auch die Bildung des Membranangriffskomplex (MAC) ein (C5b). Dabei rekrutiert der „Anker“ C5b nacheinander die Faktoren C6, C7 und C8. Der entstandene C5b678-Komplex startet dann die Polymerisierung von C9. Nach der Zusammenlagerung von bis zu 18 C9-Monomeren stellt der C5b678poly9-Komplex den fertigen Membranangriffskomplex dar, der die Zielzelle unter anderem durch Porenbildung in der Zellmembran attackiert und zu ihrer Lyse führt.[4]
Die löslichen Komplementfragmente C3a, C4a und C5a lösen eine lokale Entzündungsreaktion aus. Sie führen durch Bindung an Komplementrezeptoren der basophilen Granulozyten zu derer Freisetzung von Histamin, Heparin und Leukotrienen. Die Aktivierung von Komplementrezeptoren an Endothelzellen, glatten Muskelzellen, Monozyten, Eosinophile, und Mastzellen führt zur Bronchokonstriktion, Vasodilatation, Erhöhung der Gefäßpermeabilität und Rekrutierung (durch C5a) von Granulocyten und Monocyten an Gefässwände, was die Voraussetzung für deren Einwanderung in das Entzündungsgebiet darstellt. C3a fördert dabei vorwiegend Gewebereparatur-Mechanismen. C5a stimuliert die Entzündungsreaktion. Damit bilden die Anaphylatoxine ein wichtiges Bindeglied zwischen angeborener und adaptiver Immunabwehr.
Medizinische Bedeutung
Viele der Funktionen von Komplement sind durch das Auftreten von Krankheiten bei Defizienzen in Komplementfaktoren oder -regulatoren entdeckt worden:
- C1-Inhibitor: Ein angeborener oder erworbener C1-INH-Mangel kann zu einer übermäßigen Komplementreaktion führen, wie sie bei Auftreten von Angioödemen (Hereditäres Angioödem (HAE), Erworbenes Angioödem (AAE)) eine Rolle spielt. Durch starke Ausschüttung von Anaphylatoxinen (C3a, C5a) kommt es zur Schwellung der Atemwege, auch Haut und Darm sind betroffen.
- C2 und C4: Immunkomplex-Krankheiten treten bei Personen mit C2-Defiziten oder auch Defekten in den „frühen“ Komponenten C1q, C1r, C1s oder auch C4 auf. Ein kompletter kongenitaler C1q-Mangel ist der stärkste genetische Risikofaktor für die Entwicklung eines systemischen Lupus erythematodes (SLE).[5]
- C3: Eine Defizienz der Komponente C3 führt zum häufigen Auftreten bakterieller Infektionen (z. B. mit Neisserien).
- Fehlt aufgrund einer Mutation Faktor H, kommt es zu einer unkontrollierten Aktivierung des alternativen Weges an der Basalmembran der Nierenkörperchen und an der Bruch-Membran des Auges. Die C3-Ablagerungen führen zu einer chronischen Nierenkrankheit (Membranoproliferative Glomerulonephritis Typ II), die auch mit Sehstörungen einhergehen kann. Eine häufigere Ursache dieser Erkrankung ist ein Autoantikörper, der gegen den C3bBb-Komplex gerichtet ist, diesen stabilisiert und so den alternativen Weg aktiviert.
- Bei Defekten der sogenannten GPI-Anker auf Blutzellen können diese sich nicht mehr vor der Zerstörung durch das Komplementsystem schützen und es kommt zur paroxysmalen nächtlichen Hämoglobinurie.
Literatur
- Charles A. Janeway jr. u. a.: Immunologie. 5. Auflage. Spektrum Akademischer Verlag, Heidelberg / Berlin 2002, ISBN 3-8274-1078-9.
- Löffler, Petrides: Biochemie und Pathobiochemie. 7. Auflage. Springer-Verlag, 2004
- Siegenthaler, Blum: Klinische Pathophysiologie. 9. Auflage. Thieme-Verlag, Zürich 2006
- Prabhu Nesargikar, B. Spiller, R. Chavez: The complement system: History, pathways, cascade and inhibitors. In: European Journal of Microbiology and Immunology. Band 2, Nr. 2, Juni 2012, ISSN 2062-509X, S. 103–111, doi:10.1556/EuJMI.2.2012.2.2, PMID 24672678, PMC 3956958 (freier Volltext) – (akademiai.com [abgerufen am 28. März 2020]).
Weblinks
- Weiterführende Seite (englisch)
- Grundlagen der Immunologie 17. Vorlesung: Das Komplementsystem. Als PDF-Datei unter 16. Das Komplementsystem. Immunológiai és Biotechnológiai Intézet, UNIVERSITY OF PÉCS
Einzelnachweise
- G. J. Arlaud et al.: Structural biology of C1. In: Biochemical Society Transactions. Nr. 30, 2002, S. 1001–1006 (Artikel)., Abb.: Modulare Strukturen von C1q, C1r und C1s und makroskopisches Modell des C1 Komplexes
- C. A. Diebolder, F. J. Beurskens, R. N. de Jong, R. I. Koning, K. Strumane, M. A. Lindorfer, M. Voorhorst, D. Ugurlar, S. Rosati, A. J. Heck, J. G. van de Winkel, I. A. Wilson, A. J. Koster, R. P. Taylor, E. O. Saphire, D. R. Burton, J. Schuurman, P. Gros, P. W. Parren: Complement is activated by IgG hexamers assembled at the cell surface. In: Science Band 343, Nummer 6176, März 2014, S. 1260–1263, ISSN 1095-9203. doi:10.1126/science.1248943. PMID 24626930.
- Praveen M. Varghese, Valarmathy Murugaiah, Nazar Beirag, Nigel Temperton, Haseeb A. Khan: C4b Binding Protein Acts as an Innate Immune Effector Against Influenza A Virus. In: Frontiers in Immunology. Band 11, 2021, ISSN 1664-3224, doi:10.3389/fimmu.2020.585361 (frontiersin.org [abgerufen am 9. Februar 2022]).
- Bohana-Kashtan O., Ziporen L., Donin N., Kraus S., Fishelon Z.: Cell signals transduced by complement. In: Molecular Immunology. Nr. 41, 2004, S. 583–597, doi:10.1016/j.molimm.2004.04.007.
- Sontheimer R. et al.: C1q: Its Functions within the Innate and Adaptive Immune Responses and its Role in Lupus Autoimmunitiy. In: Journal of Investigative Dermatology. Nr. 125, 2005, S. 14–23, doi:10.1111/j.0022-202X.2005.23673.x.