Glioblastom
Das Glioblastom (auch Glioblastoma multiforme) ist der häufigste bösartige hirneigene Tumor bei Erwachsenen. Das Glioblastom weist feingewebliche Ähnlichkeiten mit den Gliazellen des Gehirns auf und wird aufgrund der sehr schlechten Prognose nach der WHO-Klassifikation der Tumoren des zentralen Nervensystems als Grad IV eingestuft. Die Behandlung besteht in operativer Reduktion der Tumormasse, Bestrahlung und Chemotherapie. Eine endgültige Heilung kann derzeit nicht erreicht werden. Die mittlere Überlebenszeit liegt bei wenigen Monaten ohne Behandlung und rund 15 Monaten bei aktuell gängigen Therapiemethoden.[1] Manche Erkrankte überleben länger,[2] nur wenige jedoch mehrere Jahre. In seltenen Fällen haben Betroffene noch über 20 Jahre gelebt.[3][4] Die Glioblastom-Zelllinie U87MG war die erste Krebszelllinie, deren Genom vollständig sequenziert wurde.
| Klassifikation nach ICD-10 | |
|---|---|
| C71 | Bösartige Neubildung des Gehirns |
| C71.0 | Zerebrum, ausgenommen Hirnlappen und Ventrikel |
| C71.1 | Frontallappen |
| C71.2 | Temporallappen |
| C71.3 | Parietallappen |
| C71.4 | Okzipitallappen |
| C71.5 | Hirnventrikel |
| C71.6 | Zerebellum |
| C71.7 | Hirnstamm |
| C71.8 | Gehirn, mehrere Teilbereiche überlappend |
| C71.9 | Gehirn, nicht näher bezeichnet |
| ICD-10 online (WHO-Version 2019) | |
Historisches
Der Begriff Glioblastoma multiforme wurde 1926 von Percival Bailey und Harvey Cushing geprägt. Die Begriffsbildung basierte auf der Vorstellung, dass sich der Tumor aus primitiven Vorstufen von Gliazellen (Glioblasten) entwickelt, sowie der Beobachtung, dass das Erscheinungsbild mit Nekrosen, Einblutungen und Zysten sehr variabel (multiform) sein kann.[5] Der von dem Pathologen Frank Burr Mallory bereits 1914 verwendete Begriff Spongioblastoma multiforme konnte sich nicht durchsetzen.[6]
Verbreitung
Glioblastome sind bei Erwachsenen die häufigsten bösartigen hirneigenen Tumore. Unter den aus dem Hirngewebe entstehenden (neuroepithelialen) Tumoren machen sie etwa die Hälfte aller Fälle aus.[7] Der Tumor tritt am häufigsten bei älteren Erwachsenen zwischen dem 60. und 70. Lebensjahr auf; das durchschnittliche Alter bei Diagnosestellung beträgt 64 Jahre. Männer sind deutlich öfter betroffen als Frauen (Verhältnis 1,7:1). Daten des amerikanischen Hirntumorregisters zeigen, dass Glioblastome bei Weißen mindestens doppelt so häufig sind wie in der schwarzen Bevölkerung. Im Vergleich zu Erwachsenen sind Glioblastome bei Kindern sehr selten. Die Inzidenz wurde in Europa und Nordamerika mit 2,9 bis 3,5 Neuerkrankungen pro Jahr auf 100.000 Einwohner ermittelt und ist in Entwicklungsländern geringer.[8][9][10] Als einziger gesicherter ursächlicher (ätiologischer) Umweltfaktor gilt derzeit eine Exposition durch ionisierende Strahlung.
Bei der Mehrzahl der Glioblastome handelt es sich um sporadisch auftretende Fälle ohne Hinweis auf eine Erblichkeit. Bei bestimmten seltenen erblichen Erkrankungen, unter anderem bei dem Li-Fraumeni-Syndrom oder dem Turcot-Syndrom, können Glioblastome jedoch in Familien gehäuft auftreten.
Krankheitsentstehung
Glioblastome können völlig neu (de novo) oder durch fortschreitende Entdifferenzierung aus weniger bösartigen Astrozytomen entstehen. Daher kommt es nicht selten vor, dass therapierte Astrozytome sich im Rezidiv als Glioblastom manifestieren. Diese sogenannten sekundären Glioblastome treten eher bei jüngeren Patienten auf und haben ein anderes Spektrum genetischer Veränderungen als neuentstandene (siehe Molekularpathologie). In einer in der Schweiz durchgeführten epidemiologischen Studie waren primäre Glioblastome im Kanton Zürich etwa zwanzigmal häufiger als sekundäre.[10]
Lokalisation
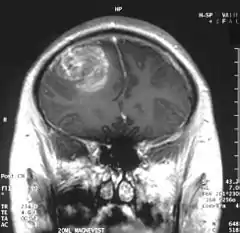
Das Glioblastom geht von der weißen Substanz aus. Seine mit Abstand häufigste Lokalisation ist das Großhirn, wo es in allen Hirnlappen entstehen kann, aber den Frontal- und den Temporallappen bevorzugt. Im Bereich von Kleinhirn, Hirnstamm und Rückenmark sind Glioblastome selten. Oft wachsen hemisphärielle Glioblastome über den Balken auf die andere Seite hinüber. Solche Tumoren werden als sogenannte „Schmetterlingsgliome“ bezeichnet. Das Wachstum von Glioblastomen ist diffus infiltrierend.
Klinische Erscheinungen
Wegen des raschen Wachstums entwickeln sich die Beschwerden meistens rasch innerhalb weniger Wochen bis Monate. Erste Symptome können anhaltende und ungewohnte Kopfschmerzen, aber auch neu auftretende epileptische Anfälle sein. Fokale neurologische Ausfälle wie Lähmungen, Aphasien und Sehstörungen können lokalisationsabhängig hinzukommen. Schließlich sind es oft auffällige Persönlichkeitsveränderungen, Apathie oder psychomotorische Verlangsamung, die den Patienten zum Arzt führen. Hirndruckzeichen wie Stauungspapille, Erbrechen, Somnolenz und Koma treten spät auf und sind prognostisch ungünstig.
Untersuchungsmethoden
Die Diagnose wird zunächst durch bildgebende Verfahren wie Computertomographie (CT) oder Magnetresonanztomographie (MRT) gestützt. In der CT-Bildgebung mit Kontrastmittel erscheint das Glioblastom unregelmäßig geformt mit randständig starker Kontrastmittelaufnahme (ringförmiges Enhancement). Bei kleineren Tumoren ist dieses ringförmig konfiguriert, bei größeren bildet es eine girlandenartige Formation aus. In der Umgebung des Tumors bildet sich typischerweise ein erhebliches Ödem aus. Der MRT-Befund ist recht typisch: die soliden Anteile des Glioblastoms reichern Kontrastmittel stark an, dagegen heben sich die Aussparungen durch zystische Anteile und die Blutungen ab. Letztendlich wird die Diagnose am Tumorgewebe, das bei einer stereotaktischen Hirnbiopsie oder Tumorresektion gewonnen wurde, neuropathologisch bestätigt. Im Einzelfall werden Supplementäruntersuchungen wie Elektroenzephalografie und Lumbalpunktion durchgeführt, die der Einschätzung der Anfallsneigung bzw. der differentialdiagnostischen Abgrenzung gegen Hirnabszesse oder Lymphome dienen.
Pathologie
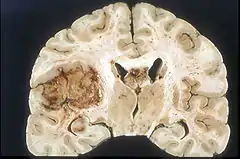
Das Glioblastom ist durch seine inhomogene und vielfältige (daher: multiforme) Erscheinung gekennzeichnet: die Tumorschnittfläche weist häufig rötliche Einblutungen und gelbliche Gewebsuntergänge (Nekrosen) auf.
Histologie
.jpg.webp)
Feingeweblich (histologisch) handelt es sich um zelldichte astrozytär differenzierte Tumoren, die diffus das umgebende reaktiv veränderte Hirngewebe infiltrieren. Die Tumorzellen sind mit multipolaren feinen Fortsätzen fibrillär-astrozytär differenziert oder weisen mit einem aufgeblähten Zytoplasma eine gemästet-zellige Differenzierung auf. Auch Riesenzellen mit bizarren Kernen oder kleinzellige Areale mit wenig ausgedehnten Zellkörpern kommen vor. Die Zellkerne sind meist chromatinreich und vielgestaltig (polymorph). Mitotische und proliferative Aktivität sind erhöht.
Entscheidend für die Diagnose des Glioblastoms (und die Abgrenzung gegenüber dem anaplastischen Astrozytom) ist nach der WHO-Klassifikation der Tumoren des zentralen Nervensystems jedoch der Nachweis von Tumornekrosen (flächenhaft oder typischerweise strichförmig mit perifokaler Zelldichtesteigerung) oder hochgradig pathologischer Blutgefäße.
Varianten
Bei Gliosarkomen handelt es sich um Glioblastome, die neben den oben beschriebenen astrozytären Tumoranteilen auch bindegewebsreiche sarkomatöse Abschnitte mit spindelzelligen Tumorzellen aufweisen. Ein Epitheloides Glioblastom weist große Epitheloidzellen mit reichlich eosinophilem Zytoplasma auf. Als Riesenzellglioblastome werden Glioblastome mit einer ausgeprägten riesenzelligen Komponente bezeichnet. Ebenfalls abzugrenzen sind Glioblastome mit oligodendroglialer Komponente, die möglicherweise eine etwas günstigere Prognose haben.[11]
Immunhistochemie
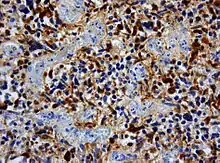
Immunhistochemisch ist in den Tumorzellen – wie in denen anderer glialer Hirntumoren – das saure Gliafaserprotein (glial fibrillary acidic protein, GFAP) nachweisbar, was in den meisten Fällen die Abgrenzung gegenüber Hirnmetastasen erlaubt.[12]
Molekularpathologie
Auf genetischer Ebene weisen Tumorzellen eines Glioblastom häufig Kopienzahlveränderungen auf, meist handelt es sich hierbei um Zugewinne auf Chromosom 7 und Verluste auf Chromosom 10. Neben zahlreichen Genmutationen finden sich in einem Drittel der Tumoren auch Genfusionen. Zusätzlich zeigen Glioblastome ein eigenständiges epigenetisches Profil. Während das epigenetische Muster beim Glioblastom relativ konstant bleibt, sind Genveränderungen im Laufe des Tumorwachstums sehr variabel.
Die Genverluste (Deletionen), die das Glioblastom ausmachen, betreffen in den meisten Fällen das Tumorsuppressor-Gen TP53 (Chromosom 17), das Retinoblastom-Suppressorgen RB-1 (Chromosom 13) und Deletionen des Chromosoms 22 sowie den Komplettverlust des langen Arms von Chromosom 10. Diese genetischen Schäden liegen häufig kombiniert vor. Bei neu entstandenen primären Glioblastomen, die überwiegend bei älteren Patienten auftreten, treten häufiger Verluste des PTEN-Gens oder eine Amplifikation des EGFR-Gens auf.[14] Bei den überwiegend im mittleren Lebensalter auftretenden sekundären Glioblastomen, welche durch eine schrittweise Fortentwicklung (Progression) aus weniger bösartigen (weniger malignen) Astrozytomen entstandenen sind, liegen häufig Mutationen des TP53-Gens vor[15]. Zudem sind Punktmutationen in für eine Isocitrat-Dehydrogenase codierenden IDH1- und IDH2-Genen in dieser Gruppe häufiger, insbesondere die R132H-Mutation im IDH1-Gen.[16][17] Die häufigste Mutation (IDH-R132H) kann zuverlässig mittels Immunhistochemie unter Anwendung eines spezifischen Antikörpers nachgewiesen werden.[18][19] Eine Mutation im IDH Gen zeigt sich vor allem für die Prognose relevant, da Patienten mit einer IDH Mutation einen Überlebensvorteil gegenüber Patienten mit der Wildtyp Variante haben[20].
Die seltenen kindlichen Glioblastome unterscheiden sich im Muster genetischer Veränderungen von den bei Erwachsenen auftretenden Tumoren: hier spielen vor allem Mutationen des H3F3A-Gens eine Rolle.[21] Anhand genetischer und epigenetischer Veränderungen wurde 2012 eine Klassifizierung der Glioblastome in sechs Untergruppen vorgeschlagen.[22]
Behandlung
Eine kurzfristige klinische Besserung kann durch Behandlung des praktisch immer vorhandenen perifokalen Hirnödems mit Corticosteroiden erreicht werden. Die neurochirurgische Operation mit Verminderung der Hauptmasse des Tumors (Tumorreduktion) kann das Fortschreiten der Erkrankung verlangsamen, aber nicht dauerhaft verhindern, da praktisch immer einzelne Tumorzellen das gesunde Gehirngewebe schon infiltrativ durchwandert haben und deswegen eine vollständige Tumorentfernung nicht möglich ist.[23] Ein innovatives Verfahren zusätzlich zur neurochirurgischen Behandlung von bösartigen Hirntumoren (z. B. dem Glioblastom) ist die fluoreszenz-gestützte Chirurgie mit 5-Aminolävulinsäure (5-ALA). Dabei erhält der Patient etwa vier Stunden vor der Operation eine körpereigene Substanz (5-ALA) als Trinklösung, die sich im Hirntumor stark anreichert und dort in einen fluoreszierenden Farbstoff umgewandelt wird. Während der Operation kann dann dieser Farbstoff durch blau-violettes Licht (Wellenlänge 410 bis 440 nm) zum Leuchten (Fluoreszenz) angeregt werden, sodass sich der Tumor (dunkelblau) vom gesunden Hirngewebe (rosa) besonders deutlich abgrenzen lässt.[24] Durch dieses Verfahren ist eine weitgehend komplette Entfernung der Tumoren viel sicherer und effektiver möglich. Das führt zu einer Verlängerung der Zeit bis zum Nachwachsen dieser Tumoren (rezidivfreies Intervall), wodurch die Prognose dieser Erkrankung deutlich verbessert wird. Das Verfahren wurde 2004 in Düsseldorf und München entwickelt und wird in vielen deutschen Kliniken angewandt. Zur Verlängerung der rezidivfreien und absoluten Überlebenszeit schließt sich an die Operation praktisch immer eine Bestrahlung und häufig auch eine Chemotherapie an, wobei insbesondere Patienten mit Nachweis epigenetischer Veränderungen (Hypermethylierung) des Promotors des DNS-Reparaturenzyms O6-Methylguanin-DNA-Methyltransferase (MGMT) von einer Chemotherapie mit Zytostatikum profitieren.[25] Bei Patienten mit neu-diagnostiziertem Glioblastom und methyliertem MGMT-Promotor wird seit 2017 in der Erstlinientherapie eine Kombination aus CCNU und Temozolomid sowie Strahlentherapie eingesetzt.[26] Weitere Chemotherapeutika, die unter anderem bei einem Rezidiv eingesetzt werden, sind Vincaalkaloide, Fotemustin und Cytarabin, wobei verschiedene Behandlungsschemata in Gebrauch sind. Welche Patienten von einer lokalen Chemotherapie mit Implantation von an Polymere gebundenem Carmustin profitieren können, ist noch unklar.[27]
Ein weiteres optionales Verfahren in der Glioblastom-Behandlung sind elektrische Wechselfelder. Dabei werden elektrische Wechselfelder in einem mittleren Frequenzbereich (200 kHz) über äußerliche Elektroden auf den erkranken Körperbereich gerichtet. So soll das Wachstum krebsartiger Tumorzellen gehemmt werden. Diese werden in der Erstlinientherapie parallel zur Chemotherapie eingesetzt.[28]
Klinische Studien
Die Entwicklung neuer Behandlungsformen bei Glioblastomen ist Gegenstand intensiver Forschung. Im Februar 2013 waren 257 klinische Studien bei Clinicaltrials.gov, einem Register der United States National Library of Medicine als aktiv oder in Vorbereitung registriert.[29] Tyrosinkinaserezeptoren, wie die Rezeptoren für epidermalen Wachstumsfaktor (EGFR) und Platelet Derived Growth Factor (PDGF), stellen mögliche Zielmoleküle für neue therapeutische Ansätze dar.[30][31]
Eine Behandlung mit Bevacizumab, einem den Vascular endothelial growth factor (VEGF) neutralisierenden Antikörper, konnte in klinischen Studien in Kombination mit dem Topoisomerasehemmer Irinotecan das Gesamtüberleben nicht verbessern, wobei einzelne Patientengruppen möglicherweise günstig auf diese Behandlung ansprechen.[32]
Die Therapie im Rahmen einer klinischen Studie mit APG101, einem vollständig humanen CD95-Fc-Fusionsprotein, das die Bindung des CD95-Liganden an den CD95-Rezeptor verhindert, stellt einen neuartigen Behandlungsansatz dar. Sie basiert auf Erkenntnissen aus dem Deutschen Krebsforschungszentrum und dem Universitätsklinikum Heidelberg, wonach in Glioblastom-Zellen die Bindung des CD95-Liganden an den CD95-Rezeptor das invasive Wachstum und die Migration der Tumorzellen stimuliert.[33] Eine Blockade dieser Bindung durch APG101 soll daher zu einer Reduktion des invasiven Wachstums und der Migration dieser Zellen führen. Eine Phase I-Studie mit 34 gesunden Probanden zur Untersuchung der Sicherheit und Verträglichkeit von APG101 zeigte eine gute Verträglichkeit der Substanz.[34] Die mögliche Wirksamkeit von APG101 wurde in einer randomisierten, kontrollierten klinischen Phase II-Studie mit Patienten untersucht, die an GBM erkrankt sind und bei denen ein Rückfall der Erkrankung aufgetreten ist. Die Patientenrekrutierung ist abgeschlossen. Insgesamt wurden 83 Patienten im Rahmen der klinischen Prüfung behandelt. Der primäre Endpunkt der Studie, eine Verdoppelung der Zahl von Patienten mit progressionsfreiem Überleben nach sechs Monaten, wurde übertroffen.[35] Während der Behandlung mit APG101, die bis zu zwei Jahre andauerte, wurden keine schwerwiegenden Nebenwirkungen beobachtet, die im Zusammenhang mit dem Wirkstoff stehen.
Auch gentherapeutische Verfahren werden im Rahmen klinischer Studien erprobt.[36]
Ein anderer experimenteller Ansatz ist die Behandlung mit Nanoteilchen.[37] Diese bestehen aus einem Eisenoxidkern sowie einer Hülle, die das Eindringen der Eisenoxidpartikel in die Krebszellen erleichtern soll. Die Partikel werden direkt in den Tumor injiziert. In mehreren Durchgängen wird der so mit den Eisenoxid-Teilchen, welche ein Ferrofluid bilden, angereicherte Tumor mit Magnetwechselfeldern auf über 46 °C erwärmt. Im Tiermodell ergaben sich deutlich verbesserte Überlebenszeiten.[38] Studienergebnisse beim Menschen liegen seit September 2010 vor,[39] seit Mitte 2011 ist die Therapie verfügbar.[40]
In einem anderen Forschungsansatz wurde wie bei anderen Krebserkrankungen mit Parvoviren gearbeitet.[41] Bis auf eine Phase I/II Studie an 18 Patienten mit Glioblastomen aus dem Jahre 2012 wurden bislang keine weiteren Daten publiziert.[42] Ein vergleichbarer Ansatz ist die Behandlung mit genetisch verändertem, attenuiertem Poliovirus (PVS-RIPO), die sich noch in einem frühen experimentellen Stadium befindet.
Prognose
Das Glioblastom ist äußerst schwierig zu behandeln. Eine endgültige Heilung ist bislang in der Regel nicht möglich. Die Behandlung mit Operation, nachfolgender Bestrahlung und Chemotherapie kann nach aktueller Studienlage die mittlere Überlebenszeit um einige Monate verlängern und die Symptome lindern. Eine Studie aus dem Jahr 2003 unterteilt die Prognose mithilfe der Recursive Partitioning Analysis (RPA) in drei Gruppen in Abhängigkeit vom Alter des Patienten, von der Art der Behandlung und vom Karnofsky-Index (KPS).[43]
| RPA Klasse | Definition | Mittlere Überlebenszeit | 1-Jahres-Überlebensrate | 3-Jahres-Überlebensrate | 5-Jahres-Überlebensrate |
|---|---|---|---|---|---|
| III | Alter < 50, KPS ≥ 90 | 17,1 Monate | 70 % | 20 % | 14 % |
| IV | Alter < 50, KPS < 90 | 11,2 Monate | 46 % | 7 % | 4 % |
| Alter > 50, KPS ≥ 70, operative Entfernung mit guter neurologischer Funktion | |||||
| V + VI | Alter ≥ 50, KPS ≥ 70, operative Entfernung mit schlechter neurologischer Funktion | 7,5 Monate | 28 % | 1 % | 0 % |
| Alter ≥ 50, KPS ≥ 70, ohne operativen Eingriff | |||||
| Alter ≥ 50, KPS < 70 |
Wegen der diffusen Infiltration des Hirngewebes durch Tumorzellen kommt es nach der Behandlung häufig innerhalb von Monaten zu einem Rezidiv. Einzelne Patienten können dessen ungeachtet mehrere Jahre bei relativ guter Gesundheit mit einem Glioblastom leben. Die Identifizierung klinischer und molekularer Faktoren, die charakteristisch für solche Langzeitüberlebenden sind, ist Gegenstand intensiver Forschung.[44]
Literatur
- Wolfgang Wick, Jörg-Christian Tonn, Michael Weller: Primäre intrakranielle und spinale Tumoren. In: Thomas Brandt, Johannes Dichgans, Hans Christoph Diener (Hrsg.): Therapie und Verlauf neurologischer Erkrankungen. 5. Auflage. Kohlhammer, Stuttgart 2007, ISBN 978-3-17-019074-0.
- Jörg-Christian Tonn, F. W. Kreth: Hirntumoren und spinale Tumoren. 4. Auflage. Zuckschwerdt-Verlag, Germering 2016, ISBN 978-3-86371-199-3.
Weblinks
- Arbeit und Struktur. Tagebuch des Schriftstellers Wolfgang Herrndorf über sein Leben mit der Diagnose Glioblastom
- Hirntumorhilfe.de: Glioblastom.
- Hoffnung trotz Glioblastom – ein Erfahrungsbericht. (PDF; 369 kB) In: Brainstorm, 01/2006, herausgegeben von der Deutschen Hirntumorhilfe
- www.glioblastom.org
- Glioblastom - Ars-Neurochirurgica.
Einzelnachweise
- Derek R. Johnson, Brian Patrick O’Neill: Glioblastoma survival in the United States before and during the temozolomide era. In: Journal of Neuro-Oncology, 2011, 107 (2), S. 359–364. doi:10.1007/s11060-011-0749-4. PMID 22045118.
- D. Krex, B. Klink, C. Hartmann, A. von Deimling, T. Pietsch, M. Simon, M. Sabel, J. P. Steinbach et al.: Long-term survival with glioblastoma multiforme. In: Brain, 130, 2007, (10), S. 2596–2606. doi:10.1093/brain/awm204.
- S. Fukushima, Y. Narita, Y. Miyakita, M. Ohno, T. Takizawa, Y. Takusagawa, M. Mori, K. Ichimura, H. Tsuda, S. Shibui: A case of more than 20 years survival with glioblastoma, and development of cavernous angioma as a delayed complication of radiotherapy. In: Neuropathology: official journal of the Japanese Society of Neuropathology. Band 33, Nummer 5, Oktober 2013, S. 576–581, doi:10.1111/neup.12022, PMID 23406431.
- F. W. Floeth, K. J. Langen, G. Reifenberger, F. Weber: Tumor-free survival of 7 years after gene therapy for recurrent glioblastoma. In: Neurology, 2003, 61, S. 270–271.
- Bailey, Cushing: Tumors of the Glioma Group. JB Lippincott, Philadelphia 1926.
- Mallory: Principles of pathologic histology. Saunders Philadelphia, 1925.
- CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2004–2006 Volltext (PDF; 14 kB)
- W. K. Cavenee et al.: Glioblastoma, in: WHO Classification of Tumours. Lyon, IARC Press, 2000.
- Amerikanisches Hirntumorregister
- H. Ohgaki, P. Kleihues: Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol 2005, 64(6):479-89; PMID 15977639.
- T. Homma, T. Fukushima u. a.: Correlation among pathology, genotype, and patient outcomes in glioblastoma. In: Journal of Neuropathology & Experimental Neurology, Band 65, Nummer 9, September 2006, S. 846–854. doi:10.1097/01.jnen.0000235118.75182.94. PMID 16957578.
- M. E. Velasco, D. Dahl u. a.: Immunohistochemical localization of glial fibrillary acidic protein in human glial neoplasms. In: Cancer. Band 45, Nummer 3, Februar 1980, S. 484–494, ISSN 0008-543X. PMID 6243508.
- D. Capper, H. Zentgraf, J. Balss, C. Hartmann, A. von Deimling: Monoclonal antibody specific for IDH1 R132H mutation. In: Acta Neuropathol.. 118, Nr. 5, November 2009, S. 599–601. doi:10.1007/s00401-009-0595-z. PMID 19798509.
- H. Ohgaki, P. Dessen u. a.: Genetic pathways to glioblastoma: a population-based study. In: Cancer Research. Band 64, Nummer 19, Oktober 2004, S. 6892–6899. doi:10.1158/0008-5472.CAN-04-1337. PMID 15466178.
- A. von Deimling, R. H. Eibl, H. Ohgaki u. a.: p53 mutations are associated with 17p allelic loss in grade II and grade III astrocytoma. In: Cancer Research. Band 52, Nummer 10, May 1992, S. 2987–2990. PMID 1349850.
- T. Watanabe, S. Nobusawa u. a.: IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. In: The American Journal of Pathology. Band 174, Nummer 4, April 2009, S. 1149–1153. doi:10.2353/ajpath.2009.080958. PMID 19246647. PMC 2671348 (freier Volltext).
- D. W. Parsons, S. Jones u. a.: An integrated genomic analysis of human glioblastoma multiforme. In: Science, Band 321, Nummer 5897, September 2008, S. 1807–1812. doi:10.1126/science.1164382. PMID 18772396. PMC 2820389 (freier Volltext).
- M. Preusser, A. Wöhrer, S. Stary, R. Höftberger, B. Streubel, J. A. Hainfellner: Value and limitations of immunohistochemistry and gene sequencing for detection of the IDH1-R132H mutation in diffuse glioma biopsy specimens. In: J Neuropathol Exp Neurol., 2011 Aug, 70(8),715–723. doi:10.1097/NEN.0b013e31822713f0.
- D. Capper, S. Weissert, J. Balss, A. Habel, J. Meyer, D. Jäger, U. Ackermann, C. Tessmer, A. Korshunov, H. Zentgraf, C. Hartmann, A. von Deimling: Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. In: Brain Pathol., 2010 Jan, 20 (1), S. 245–254. doi:10.1111/j.1750-3639.2009.00352.x.
- Glioblastom. Abgerufen am 13. August 2020.
- J. Schwartzentruber, A. Korshunov, X. Y. Liu, D. T. Jones, E. Pfaff, K. Jacob, D. Sturm, A. M. Fontebasso, D. A. Quang, M. Tönjes, V. Hovestadt, S. Albrecht, M. Kool, A. Nantel, C. Konermann, A. Lindroth, N. Jäger, T. Rausch, M. Ryzhova, Jan Korbel, T. Hielscher, P. Hauser, M. Garami, A. Klekner, L. Bognar, M. Ebinger, M. U. Schuhmann, W. Scheurlen, A. Pekrun, M. C. Frühwald, W. Roggendorf, C. Kramm, M. Dürken, J. Atkinson, P. Lepage, A. Montpetit, M. Zakrzewska, K. Zakrzewski, P. P. Liberski, Z. Dong, P. Siegel, A. E. Kulozik, M. Zapatka, A. Guha, D. Malkin, J. Felsberg, G. Reifenberger, A. von Deimling, K. Ichimura, V. P. Collins, H. Witt, T. Milde, O. Witt, C. Zhang, P. Castelo-Branco, P. Lichter, D. Faury, U. Tabori, C. Plass, J. Majewski, S. M. Pfister, N. Jabado: Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. In: Nature. Band 482, Nummer 7384, Februar 2012, S. 226–231. doi:10.1038/nature10833. PMID 22286061.
- D. Sturm, H. Witt, V. Hovestadt, D. A. Khuong-Quang, D. T. Jones, C. Konermann, E. Pfaff, M. Tönjes, M. Sill, S. Bender, M. Kool, M. Zapatka, N. Becker, M. Zucknick, T. Hielscher, X. Y. Liu, A. M. Fontebasso, M. Ryzhova, S. Albrecht, K. Jacob, M. Wolter, M. Ebinger, M. U. Schuhmann, T. van Meter, M. C. Frühwald, H. Hauch, A. Pekrun, B. Radlwimmer, T. Niehues, G. von Komorowski, M. Dürken, A. E. Kulozik, J. Madden, A. Donson, N. K. Foreman, R. Drissi, M. Fouladi, W. Scheurlen, A. von Deimling, C. Monoranu, W. Roggendorf, C. Herold-Mende, A. Unterberg, C. M. Kramm, J. Felsberg, C. Hartmann, B. Wiestler, W. Wick, T. Milde, O. Witt, A. M. Lindroth, J. Schwartzentruber, D. Faury, A. Fleming, M. Zakrzewska, P. P. Liberski, K. Zakrzewski, P. Hauser, M. Garami, A. Klekner, L. Bognar, S. Morrissy, F. Cavalli, M. D. Taylor, P. van Sluis, J. Koster, R. Versteeg, R. Volckmann, T. Mikkelsen, K. Aldape, G. Reifenberger, V. P. Collins, J. Majewski, A. Korshunov, P. Lichter, C. Plass, N. Jabado, S. M. Pfister: Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. In: Cancer Cell, Band 22, Nummer 4, Oktober 2012, S. 425–437. doi:10.1016/j.ccr.2012.08.024. PMID 23079654.
- Glioblastom. Ars Neurochirurgica
- B. Pakrah-Bodingbauer, M. Loyoddin, S. Oberndorfer, G. Kleinpeter: Wertigkeit der 5-Aminolävulinsäure- (5-ALA-)gestützten Gliomchirurgie. (PDF; 767 kB) In: J Neurol Neurochir Psychiatr. Band 10, Nummer 2, 2009, S. 22–25.
- R. Stupp, W. P. Mason u. a.: Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. In: The New England journal of medicine. Band 352, Nummer 10, März 2005, S. 987–996. doi:10.1056/NEJMoa043330. PMID 15758009.
- Ulrich Herrlinger, Theophilos Tzaridis, Frederic Mack, Joachim Steinbach, Uwe Schlegel: ACTR-58. Phase III Trial of CCNU/Temozolomide (TMZ) Combination Therapy vs. Standard TMZ Therapy for Newly Diagnosed MGMT-Methylated Glioblastoma Patients: The CeTeg/NOA-09 trial. In: Neuro-Oncology. Band 19, suppl_6, November 2017, ISSN 1522-8517, S. vi13–vi14, doi:10.1093/neuonc/nox168.049.
- J. Perry, A. Chambers u. a.: Gliadel wafers in the treatment of malignant glioma: a systematic review. In: Current Oncology Band 14, Nummer 5, Oktober 2007, S. 189–194. PMID 17938702. PMC 2002480 (freier Volltext).
- Roger Stupp, Sophie Taillibert, Andrew Kanner, William Read, David M. Steinberg: Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. In: JAMA. Band 318, Nr. 23, 19. Dezember 2017, ISSN 0098-7484, S. 2306, doi:10.1001/jama.2017.18718, PMID 29260225, PMC 5820703 (freier Volltext) – (jamanetwork.com [abgerufen am 6. Juli 2020]).
- clinicaltrials.gov Abfrage bei clinicaltrials.gov am 24. Februar 2013.
- I. K. Mellinghoff, M. Y. Wang, I. Vivanco, D. A. Haas-Kogan, S. Zhu, E. Q. Dia, K. V. Lu, K. Yoshimoto, J. H. Huang, D. J. Chute, B. L. Riggs, S. Horvath, L. M. Liau, W. K. Cavenee, P. N. Rao, R. Beroukhim, T. C. Peck, J. C. Lee, W. R. Sellers, D. Stokoe, M. Prados, T. F. Cloughesy, C. L. Sawyers, P. S. Mischel: Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. In: The New England Journal of Medicine. Band 353, Nummer 19, November 2005, S. 2012–2024. doi:10.1056/NEJMoa051918. PMID 16282176.
- D. A. Reardon, M. J. Egorin, J. A. Quinn, J. N. Rich, J. N. Rich, S. Gururangan, I. Gururangan, J. J. Vredenburgh, A. Desjardins, S. Sathornsumetee, J. M. Provenzale, J. E. Herndon, J. M. Dowell, M. A. Badruddoja, R. E. McLendon, T. F. Lagattuta, K. P. Kicielinski, G. Dresemann, J. H. Sampson, A. H. Friedman, A. J. Salvado, H. S. Friedman: Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. In: Journal of Clinical Oncology. Band 23, Nummer 36, Dezember 2005, S. 9359–9368. doi:10.1200/JCO.2005.03.2185. PMID 16361636.
- S. Sathornsumetee, Y. Cao, J. E. Marcello, J. E. Herndon, R. E. McLendon, A. Desjardins, H. S. Friedman, M. W. Dewhirst, J. J. Vredenburgh, J. N. Rich: Tumor angiogenic and hypoxic profiles predict radiographic response and survival in malignant astrocytoma patients treated with bevacizumab and irinotecan. In: Journal of clinical oncology. Band 26, Nummer 2, Januar 2008, S. 271–278. doi:10.1200/JCO.2007.13.3652. PMID 18182667.
- S. Kleber, I. Sancho-Martinez u. a.: Yes and PI3K bind CD95 to signal invasion of glioblastoma. In: Cancer cell. Band 13, Nummer 3, März 2008, S. 235–248. doi:10.1016/j.ccr.2008.02.003. PMID 18328427.
- Tuettenberg et al.: Pharmacokinetics, pharmacodynamics, safety and tolerability of APG101, a CD95-Fc fusion protein, in healthy volunteers and two glioma patients. In: International Immunopharmacology Nummer 13, Mai 2012, S. 93–100
- Apogenix drug candidate APG101 reaches primary goal in Phase II brain cancer trial.
- P. Dent, A. Yacoub u. a.: Searching for a cure: gene therapy for glioblastoma. In: Cancer Biology and Therapy, Band 7, Nummer 9, September 2008, S. 1335–1340, ISSN 1555-8576. PMID 18708757. (Review).
- K. Maier-Hauff, R. Rothe u. a.: Intracranial thermotherapy using magnetic nanoparticles combined with external beam radiotherapy: results of a feasibility study on patients with glioblastoma multiforme. In: Journal of neuro-oncology. Band 81, Nummer 1, Januar 2007, S. 53–60, ISSN 0167-594X. doi:10.1007/s11060-006-9195-0. PMID 16773216.
- A. Jordan, R. Scholz u. a.: The effect of thermotherapy using magnetic nanoparticles on rat malignant glioma. In: Journal of neuro-oncology. Band 78, Nummer 1, Mai 2006, S. 7–14, ISSN 0167-594X. doi:10.1007/s11060-005-9059-z. PMID 16314937.
- Bekanntgabe klinischer Studienergebnisse. MagForce AG Berlin, 21. September 2010.
- Nanotherm-Therapie bei Rückfällen von Gehirntumoren. (Memento des Originals vom 23. September 2015 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. Charité Universitätsmedizin Berlin, vom 7. Juli 2011
- Viren gegen Krebs: Bösartige Hirntumoren bilden sich nach Therapie mit Parvoviren vollständig zurück. (PDF; 39 kB) Deutsches Krebsforschungszentrum, Pressemitteilung vom 3. Mai 2010
- Antonio Marchini, Serena Bonifati u. a.: Oncolytic parvoviruses: from basic virology to clinical applications. In: Virology Journal, 12, 2015, S. 6, doi:10.1186/s12985-014-0223-y.
- E. G. Shawl, W. Seiferheld, C. Scott u. a.: Re-examining the radiation therapy oncology group (RTOG) recursive partitioning analysis (RPA) for glioblastoma multiforme (GBM) patients. In: International Journal of Radiation Oncology – Biology – Physics. 57, Nr. 2, 2003, S. 135–136. doi:10.1016/S0360-3016(03)00843-5.
- D. Krex, B. Klink u. a.: Long-term survival with glioblastoma multiforme. In: Brain, Band 130, 2007, S. 2596–2606. doi:10.1093/brain/awm204. PMID 17785346. (Review).