Astrozytom
Astrozytome gehören zu den häufigsten Tumoren des Gehirns und treten vorwiegend im mittleren Lebensalter auf. Sie haben ihren Ursprung in den Astrozyten, die zum Stützgewebe (Gliazellen) des Zentralnervensystems gehören, und werden deshalb den Gliomen zugeordnet. Der Grad der Bösartigkeit wird nach einer Gewebeprobe mikroskopisch anhand feststehender Kriterien bestimmt.
| Klassifikation nach ICD-10 | |
|---|---|
| D33 | Gutartige Neubildung des Gehirns und Zentralnervensystems |
| D33.0 | Gehirn, supratentoriell |
| D33.1 | Gehirn, infratentoriell |
| D33.2 | Gehirn, nicht näher bezeichnet |
| D33.3 | Hirnnerven |
| D33.4 | Rückenmark |
| D33.7 | Sonstige Teile des Zentralnervensystems |
| D33.9 | Zentralnervensystem, nicht näher bezeichnet |
| D43 | Neubildung unsicheren oder unbekannten Verhaltens des Gehirns und des Zentralnervensystems |
| D43.0 | Gehirn, supratentoriell |
| D43.1 | Gehirn, infratentoriell |
| D43.2 | Gehirn, nicht näher bezeichnet |
| D43.3 | Hirnnerven |
| D43.4 | Rückenmark |
| D43.7 | Sonstige Teile des Zentralnervensystems |
| D43.9 | Zentralnervensystem, nicht näher bezeichnet |
| C71 | Bösartige Neubildung des Gehirns |
| C71.0 | Zerebrum, ausgenommen Hirnlappen und Ventrikel |
| C71.1 | Frontallappen |
| C71.2 | Temporallappen |
| C71.3 | Parietallappen |
| C71.4 | Okzipitallappen |
| C71.5 | Hirnventrikel |
| C71.6 | Zerebellum |
| C71.7 | Hirnstamm |
| C71.8 | Gehirn, mehrere Teilbereiche überlappend |
| C71.9 | Gehirn, nicht näher bezeichnet |
| C72 | Bösartige Neubildung des Rückenmarkes, der Hirnnerven und anderer Teile des Zentralnervensystems |
| C72.0 | Rückenmark |
| C72.1 | Cauda equina |
| C72.2 | Nn. olfactorii (I. Hirnnerv) |
| C72.3 | N. opticus (II. Hirnnerv) |
| C72.4 | N. vestibulocochlearis (VIII. Hirnnerv) |
| C72.5 | Sonstige und nicht näher bezeichnete Hirnnerven |
| C72.8 | Gehirn und andere Teile des Zentralnervensystems, mehrere Teilbereiche überlappend |
| C72.9 | Zentralnervensystem, nicht näher bezeichnet |
| ICD-10 online (WHO-Version 2019) | |
Einleitung
Bislang wurden die unterschiedlichsten tumorauslösenden Reize (Onkogene) für Astrozytome vorgeschlagen. Hiervon haben über 70 % der weniger differenzierten (WHO Grad II und III) eine Veränderung des Erbgutes für das zytosolische Enzym Isocitrat-Dehydrogenase (IDH). Für undifferenzierte Tumoren (WHO Grad IV) – Glioblastome – liegt diese Rate der Erbgutveränderung (Mutation) bei 100 %.[1] Bei der IDH-Mutation ist dann eine einzige Aminosäure an Position 132 – Arginin gegen Histidin – ausgetauscht. Diese IDH1-R132H-Mutation beeinträchtigt die üblicherweise erfolgende Umwandlung von Isocitrat in alpha-Ketoglutarat. Stattdessen erfolgt eine direkte Reduktion von alpha-Ketoglutarat in 2-Hydroxyglutarat, dessen erhöhter Spiegel ein Risiko für die Entstehung von Hirntumoren ist.[2] Daneben wurde die Exposition gegenüber erdölbezogenen Chemikalien (Petrochemicals) und Strahlung von Mobiltelefonen als Risikofaktor angesehen, dies konnte jedoch in entsprechenden Studien nicht bestätigt werden.[3][4] Die Krankheitszeichen sind von der Lage des Tumors im Gehirn oder im Wirbelkanal abhängig. In der Regel beginnen sie mit andauernden Schmerzen, Empfindungsstörungen oder auch einer Epilepsie. Später treten oft noch neurologische Ausfälle (beispielsweise Lähmungen) der betroffenen Regionen hinzu. Der Nachweis erfolgt mittels Kontrastmittel im CT oder einer Kernspintomografie und um entsprechende Zellen zu erhalten, durch eine Biopsie. Die Behandlung zielt auf eine möglichst komplette chirurgische Entfernung des Tumors ab. Bei den höhergradigen Tumoren kann eine anschließende Bestrahlung des Tumorbereiches notwendig sein. Eine Heilung durch alleinige Chemotherapie, z. B. mit Temozolomid, ist zurzeit (2009) nicht möglich. Diese kann in Kombination mit der Strahlentherapie jedoch die Überlebenswahrscheinlichkeit verbessern.
Niedrig-maligne Astrozytome
Das Niedrig-maligne Astrozytom (Grad II WHO) (Synonym: englisch Low Grade Astrocytoma) ist ein Tumor des Gehirns, der von einer bestimmten Zellart des Nervensystems (den Astrozyten) ausgeht und damit zu den sogenannten Gliomen gehört. Betroffen sind vorwiegend junge Erwachsene, bei denen die Erkrankung meist mit einem erstmaligen epileptischen Anfall auffällig wird. Die Untersuchungsbefunde bei einem Astrozytom ähneln denen eines ischämischen Hirninfarktes. Die Therapie besteht in der operativen Entfernung des Tumors, gegebenenfalls mit anschließender Bestrahlung. Die 5-Jahres-Überlebensrate der Patienten liegt bei 40 bis 50 %.
Allgemeine Betrachtung

Astrozytome sind Hirntumoren, bei denen zunächst nicht von Bösartigkeit ausgegangen wird. Wie viele Tumoren verursachen sie zu Beginn der Erkrankung keine Beschwerden. Sie können sogar sehr groß werden, bevor das erste Mal Beschwerden auftreten. Häufig werden sie deshalb erst in fortgeschrittenen Stadium entdeckt, können dann aber immer noch gutartig sein. Oft fällt der Tumor bei einer Computertomographie auf, die wegen eines erstmaligen epileptischen Anfalls veranlasst wurde. Die Diagnose „Astrozytom“ kann jedoch erst durch eine Gewebeentnahme (Biopsie) aus der entsprechenden Gehirnregion gesichert werden.
Die Therapieplanung beginnt mit der Frage, ob der zunächst gutartige Tumor überhaupt behandelt werden soll. Angesichts der räumlichen Enge in der Schädelhöhle sind Größe und Wachstum des Tumors von großer Bedeutung. Da es keine wirksamen Medikamente gegen Astrozytome gibt (die Chemotherapie ist nicht wirksam) und eine Bestrahlung nur in bestimmten Fällen hilft, bleibt oft nur die Operation. Häufig wird versucht, das Astrozytom operativ zu entfernen. Eine Operation wird meist wiederum nicht sofort nach der ersten Diagnosestellung als notwendig erachtet.
Die Notwendigkeit zur Operation hängt mit der Neigung der Astrozytome zusammen, sich zu bösartigen Formen weiterzuentwickeln. Es gibt starke Hinweise darauf, dass sich Astrozytome zunächst zu sogenannten anaplastischen Astrozytomen (Astrozytom Grad III WHO, auf dem Weg zur Bösartigkeit schon weiter fortgeschrittene Tumoren) und schließlich zu Glioblastomen (Astrozytom Grad IV WHO, sehr bösartiger Hirntumor) entwickeln können. Bei einem Teil der Patienten scheint schon früh festzustehen, ob sich ein Astrozytom zu einem bösartigen Tumor entwickeln wird. In diesen Fällen ist die Prognose unabhängig von der Behandlungsmethode schlecht. Ein weiterer Teil der Patienten wird kein Glioblastom entwickeln, bei ihnen ist die Prognose gut, egal ob früher oder später operiert wird. Wie sich ein Astrozytom entwickelt, lässt sich nicht vorhersagen.
Epidemiologie
Die durchschnittliche jährliche Inzidenzrate (Neuerkrankungen) der niedrig-malignen Astrozytome beträgt 0,9 auf 100.000 Einwohner. Das mittlere Alter der Patienten mit diesem Tumor liegt bei 35 Jahren. 55 bis 65 % der Patienten sind Männer. Es gibt keine Häufung der Astrozytome innerhalb ethnischer Gruppen. Patienten mit einer Phakomatose (erbliche Tumorerkrankung mit Fehlbildungen der Haut und des Nervensystems) haben ein erhöhtes Risiko, an einem Astrozytom zu erkranken. Bei der Neurofibromatose Typ 1 findet man gehäuft Optikus-Gliome (Tumoren des Sehnerves). Astrozytome stellen 15 % der Gliome in Hirnstamm, Großhirnrinde und Kleinhirn dieser Patienten. Patienten mit tuberöser Sklerose erleiden in 5 % der Fälle im Jugendalter subependymale Riesenzellastrozytome im Bereich des Foramen Monroi.
Bevorzugte Orte der Tumorlokalisation
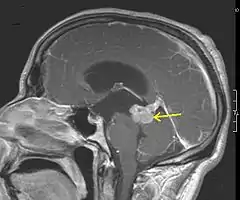
Astrozytome finden sich vorwiegend im Bereich der Konvexität (äußere Bereiche des Großhirns) und dort im Frontallappen und im Temporallappen. Die Tumoren entwickeln sich im Bereich der weißen Substanz (Nervenzellfaserbündel) der Hemisphären (Hirnhälften) und liegen meist „unterhalb“ der Hirnrinde (subkortikal). Niedrigmaligne Gliome können aber auch in allen anderen Abschnitten des Gehirns und des Rückenmarks auftreten.
Symptome
Das bei weitem häufigste erste klinische Symptom bei über 50 % der Patienten ist ein epileptischer Anfall. Der Mechanismus der Symptome ist die Infiltration und Zerstörung benachbarter Neurone. Durch einen Verdrängungsdruck kommt es zum „Hirndruck“. Das häufigste gemeinsame Zeichen dieser Mechanismen ist ein Papillenödem (Vorwölbung der Papille, der Austrittsstelle des Sehnerven in der Netzhaut, ohne Minderung der Sehkraft). Kopfschmerzen, Lethargie und Persönlichkeitsveränderungen sind ebenfalls häufige Zeichen eines beginnenden Hirndruckes. Fokale neurologische Zeichen (Lähmung, Störung der Hirnnervenfunktion, Kopfschmerzen) gehen der Diagnosestellung oft Jahre voraus.
Technische Untersuchungsbefunde
Die technischen Untersuchungsbefunde eines Astrozytoms gleichen denen eines ischämischen Hirninfarktes:
Die craniale Computertomographie (CCT) ohne Kontrastmittel zeigt gelegentlich unscharfe Hypodensitäten, manchmal ein „Marklagerödem“, aber auch „zystische“ Formationen. Mit Kontrastmittel sind meist runde, hochparietale oder frontotemporale Hypodensitäten mit lokalem Masseneffekt ohne Anreicherung von Kontrastmittel („Infarktareale“) erkennbar. Patienten, bei denen es zu einer Kontrastmittelanreicherung im Tumor kommt, haben ein siebenmal höheres Risiko für ein Rezidiv.
Der Liquorbefund ist bei Patienten mit einem Astrozytom in der Regel normal.
In der Hirnangiographie zeigen Astrozytome typischerweise keine pathologische Blutgefäßarchitektur (Vaskularisierung).
In der Kernspintomographie sieht man üblicherweise in der T1-Sequenz homogene Hypointensitäten und in der T2-Sequenz homogene Hyperintensitäten. Im Allgemeinen findet man keine Nekrosen, keine Blutung und keine Kontrastmittelanreicherung. Vereinzelt sind in der T1-Wichtung pathologisch strukturierte isointense Formationen erkennbar.
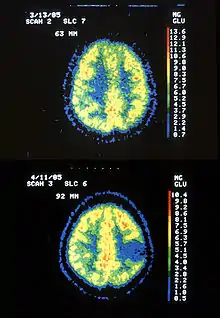
Im PET-Scan des Glucose-Stoffwechsels (FDG-PET) stellt sich das Astrozytom hypometabolisch dar („kalter Knoten“, das heißt, es ist Gewebe mit vermindertem Stoff- und Energieumsatz). Entdifferenzierungen innerhalb des Tumors führen gelegentlich zu malignen Zwischenstufen, die dann im PET-Bild als „hot spots“ (Gewebe mit erhöhtem Stoff- und Energieumsatz) innerhalb des „kalten Knotens“ erscheinen können.
Pathologie
Pathologen unterscheiden drei Formen von niedriggradigen Astrozytomen WHO Grad II: Das protoplasmatische Astrozytom, das fibrilläre Astrozytom und das gemistozytäre Astrozytom.
Mit bloßem Auge betrachtet (makroskopisch) erscheint das protoplasmatische Astrozytom als eine weiche graue Cortexexpansion. Der Tumor geht ohne genaue Grenze in Cortex und Marklager über und zeigt manchmal zystische Formationen im Schnittbild. Im Gegensatz dazu scheinen die fibrillären Astrozytome von festerer Gewebekonsistenz zu sein.
Bei mikroskopischer Beurteilung zeigen die sehr seltenen, zellarmen protoplasmatischen Astrozytome eine gleichmäßige Verteilung der Tumorzellen in einer mit Eosin anfärbbaren Matrix. Die Tumorzellen sind zart bis plump und arm an Fortsätzen. Mikrozysten kommen vor. Man sieht wenig Blutgefäße, welche unauffällig konfiguriert sind. Die am häufigsten vorkommenden fibrilläre Astrozytom zeigen eine eher lockere Durchsetzung des Gewebes mit Gliafasern. Diese lassen sich mit Antikörpern gegen das saure Gliafaserprotein (GFAP) darstellen. In den meisten Neuropathologien wird unterdessen nicht mehr zwischen protoplasmatischen und fibrillären Astrozytomen WHO Grad II differenziert, da es sich wahrscheinlich um die gleiche Tumorentität handelt. Das gemistozytäre oder gemästetzellige Astrozytom weist Tumorzellen mit großen Zytoplasmata und teils mehreren exzentrisch gelegenen Kernen auf. Man sieht in WHO Grad II Astrozytomen keine Mitosen. Allerdings rechtfertigt der Nachweis einer einzigen Mitose noch nicht die Diagnose eines anaplastischen Astrozytoms WHO Grad III. Das allgemeine mikroskopische Bild eines Astrozytoms kann folgendermaßen beschrieben werden: Vorbestehende Blutgefäße werden verdrängt, das infiltrierte Gewebe im Randbereich ist gut erhalten, die Hirnhäute können infiltriert sein und der Tumor kann zum Beispiel eine Gewebebrücke durch die Sylvische Fissur bilden. Eine Liquoraussaat von Tumorzellen ist selten. Selten gibt es degenerative Veränderungen innerhalb von Mikrozysten mit Verkalkungen.
Der wichtigste immunhistochemische Befund ist das GFA-Protein, welches von Tumorzellen und deren Fortsätzen gebildet wird. Weiterhin sollte die Proliferationsrate mittels Mib1-spezifischen Antikörpern bestimmt werden.
Die Elektronenmikroskopie hat keine Bedeutung in der Diagnostik glialer Tumoren. In der Elektronenmikroskopie sieht man Intermediärfilamente mit einer Größe von 7 bis 11 nm im Zellplasma. Mikrotubuli finden sich in manchen Zellfortsätzen.
Maligne Transformation des Tumors
Die Frage nach der Umwandlung in eine bösartige Form ist wichtig, da sie die Klassifikation und die Prognose dieser Tumorerkrankungen betrifft. Man findet in Gewebeproben resezierter Astrozytome nicht selten kleine anaplastische Foci, Areale mit höhermalignen Tumorzellpopulationen. Im Folgenden sind dazu die wichtigsten klinischen Studien in Kurzform dargestellt:
- Scherer[5] fand 1940 als Erstautor in 13 von 18 Fällen Anaplasien.
- Russell und Rubinstein[6] beschrieben 1989 in 55 Autopsien 50 % anaplastische Foci. Dieselben Autoren fanden in 129 Autopsien von Glioblastoma multiforme in ca. 30 % der Fälle Hinweise für eine Genese aus Astrozytomen.
- Müller et al.[7] untersuchten 1977 72 Patienten mit der initialen Diagnose Astrozytom. Zum Zeitpunkt eines Rezidiv zeigten 15 % der Patienten eine unveränderte Pathologie, 55 % anaplastische Astrozytome und 30 % multiforme Glioblastome. Die durchschnittliche Dauer zwischen Erstdiagnose und Rezidiv war 2,5 Jahre.
- Laws et al.[8] fanden 1984 bei 79 Patienten mit rezidivierendem Tumorwachstum eine Dedifferenzierung zu höhergradigen Astrozytomformen in 50 % der Fälle.
- Piepmeier[9] hingegen fand lediglich bei 13 % der untersuchten Patienten bei einem Tumor-Rezidiv oder Autopsie eine maligne Transformation. Allerdings war die durchschnittliche Zeit zur Nachuntersuchung mit 5 Jahren recht kurz.
Zusammenfassend kann man sagen, dass der Nachweis anaplastischer Herde zum Zeitpunkt einer zweiten Resektion nicht notwendigerweise das Resultat einer initialen negativen Selektion darstellt. Oder einfach ausgedrückt: Gutartige Astrozytome verwandeln sich mit großer Wahrscheinlichkeit im Laufe der Zeit in bösartige Tumoren.
Zur Frage der Ursachen der malignen Transformation liegen folgende Befunde vor. Beim Übergang vom niedrig-malignen Astrozytom über das anaplastische Astrozytom zum Glioblastom zeigt das Astrozytom in keinem der untersuchten Fälle p53-Mutationen, das anaplastische Astrozytome 36 % p53-Mutationen und Glioblastome 28 % p53-Mutationen. Man findet außerdem eine deutliche Zunahme von Anomalien des Chromosom 10: bei Astrozytom Grad I keine Anomalien, beim anaplastischen Astrozytom 23 % Anomalien und beim Glioblastom 61 % Anomalien.
Leitlinien für die Diagnosestellung
Die Diagnose eines Astrozytoms kann nicht klinisch oder durch technische Untersuchungsverfahren gestellt werden. Die einzige Möglichkeit ein Astrozytom zu diagnostizieren, ist eine feingewebliche Untersuchung des suspekten Gewebes. Ein Astrozytom unterscheidet sich in der Bildgebung im Zweifelsfalle nicht von einem Hirninfarktareal. Deshalb wird es aus Sicht der Neurologie als vorteilhaft betrachtet, dass Ärzte nicht aufgrund der technischen Befunde urteilen, sondern aufgrund der Anamnese: ein junger Mensch mit einem Astrozytom hat kein hirninfarktähnliches Ereignis, das etwa einer Lähmung vorangegangen ist. Die Lähmung kam nicht plötzlich, sondern langsam. Wenn also die technischen Befunde (CCT, MRT usw.) wie ein Hirninfarkt aussehen, die Schilderung eines Patienten aber dazu nicht passt und keine Gefäßrisikofaktoren vorliegen, wird der Neurologe an einen Tumor denken und eine Hirnbiopsie empfehlen.
Therapie
Bei der Behandlung der Astrozytome steht die operative Entfernung des Tumors im Vordergrund. Das generelle Konzept für einen Therapieplan von Astrozytom-Patienten ist allerdings umstritten. Die erste Regel für jede Tumorchirurgie lautet, so früh wie möglich zu operieren. Da allerdings durch verbesserte bildgebende Verfahren zunehmend Diagnosestellungen erfolgen, bevor Patienten neurologische Ausfälle erlitten haben, wurde vorgeschlagen, die Operation in Fällen, bei denen sie zu einem postoperativen neurologischen Defizit führen würde, zu verschieben, bis der Tumor radiologische Veränderungen zeigt. Diese Empfehlung wurde angesichts der Tatsache gemacht, dass eine Verlängerung der Lebenserwartung von Astrozytom-Patienten durch eine frühzeitige Operation nicht bewiesen ist.
Recht[10] verglich 1992 26 Patienten mit Astrozytomen und nachfolgend verzögerter Operation mit 20 Patienten mit unmittelbar nach Diagnosestellung folgender Operation. Es fand sich kein signifikanter Unterschied, weder im Ausmaß der Tumor-Dedifferenzierung noch bei der Lebenserwartung. In der abschließenden Zusammenfassung hieß es: „deferring surgery will not make worse outcome“ – „Abwarten verschlechtert nicht die Prognose“.
Nach Guthrie und Laws (1990) sollte dabei das Tumorzentrum gesucht werden und dann nach peripher reseziert werden. Dies kann CT- oder MRI-gesteuert stereotaktisch erfolgen. Stereotaktische Resektionen kleiner Tumoren oder stereotaktische Biopsien werden heute praktisch ambulant vorgenommen.
Es gibt keine randomisierte, kontrollierte und prospektive klinische Studie zur Frage einer postoperativen Strahlentherapie von Astrozytom-Patienten. Kaum zwei der publizierten Studien sind auch nur in einzelnen Aspekten der Auswahl der Patienten, Alter, Ausmaß oder Lokalisation des Tumors, pathologischer Klassifikation, Strahlendosis der bestrahlten Patienten etc. miteinander vergleichbar.
- Bouchard und Peirce[11] zeigten 1960, dass bei 81 Astrozytom-Patienten mit einer postoperativen Bestrahlung gegenüber 71 Astrozytom-Patienten ohne Bestrahlung die 3-Jahres-Überlebensrate gleich war (62 % gegenüber 59 %), aber die 5 Jahres-Überlebensrate der bestrahlten Gruppe verbessert war (49 % gegenüber 38 %).
- Gol[12] berichtete ähnliches in einer Studie mit 194 Astrozytom-Patienten.
- Uihlein et al.[13] dokumentierten 1966 das Gegenteil in einer Studie der Mayo-Klinik.
- Garcia et al.[14] berichten retrospektiv von 86 Patienten über einen Zeitraum von 1950 bis 1979: 3-Jahresrate bestrahlt/nichtbestrahlt: 61 %/35 %. 5-Jahresrate bestrahlt/nichtbestrahlt 40 %/22 %. 10-Jahresrate bestrahlt/nichtbestrahlt 9 %/9 %.
Trotz der prinzipiell erheblichen Mängel aller durchgeführten Studien kommt die Mehrzahl der im englischsprachigen Raum publizierten Studien zu einem Vorteil durch eine postoperative Bestrahlung von Astrozytom-Patienten. Komplikationen ergeben sich aus der Strahlennekrose des Hirngewebes. Diese tritt vor allem bei Ganzkopfbestrahlungen auf.
Es existieren keine Studien, die einen Nutzen für die Patienten durch eine Chemotherapie des Astrozytoms belegen. Im Allgemeinen gilt, dass die Chemotherapie niedriggradiger Astrozytome keine große Bedeutung hat,[15] bzw. selten indiziert ist.[16]
Die Hauptursache für ein Therapieversagen ist ein lokales Rezidiv. Wenn eine erneute Therapie erforderlich ist, ist der erste Schritt eine Biopsie. Bei weiterhin bestehendem Astrozytom erfolgen radiologische Kontrollen. Bei weiterem Wachstum des Tumors wird eine Resektion notwendig werden. Bei maligner Transformation ist eine aggressivere Therapie notwendig. Die Zweitbestrahlung eines Astrozytom-Rezidivs ist bisher ein experimentelles Verfahren.
Prognose
Die 5-Jahres-Überlebensrate von Patienten mit einem Astrozytom beträgt 40 bis 50 %. Die 10-Jahres-Überlebensrate liegt bei 20 bis 30 %. Die jüngsten Daten zu dieser Frage zeigen eine leichte Verbesserung der Prognose: die 5-Jahres-Überlebensrate stieg demnach auf 65 % und die 10-Jahres-Überlebensrate auf 40 %.
Astrozytome bei Kindern
Astrozytome sind der häufigste Tumor mit hemisphärischer Lokalisation (Großhirnrinde) im Kindesalter. Sie machen 8 % aller pädiatrischen intrakraniellen Neoplasien aus. Der Altersgipfel ihres Auftretens liegt zwischen 8 und 12 Jahren. Das Geschlecht spielt keine Rolle: Jungen und Mädchen sind in etwa gleich häufig betroffen. Neben der Lokalisation im Großhirn können Astrozytome auch im Rückenmark und entlang der Hirnnerven vorkommen. Besonders häufig sind Astrozytome am Sehnerven (Nervus Opticus), dort oft bei Patienten mit einer Neurofibromatose. Das Vorkommen von Astrozytomen ist aber auch bei anderen Phakomatosen gehäuft (siehe oben).
Der häufigste Hirntumor im Kindesalter ist das Medulloblastom.
Klinische Symptome sind Kopfschmerz, Schwindel, Erbrechen, Krampfanfälle und Sehstörungen, wie z. B. spontanes Schielen, gelegentlich ein fokales neurologisches Defizit (beispielsweise eine Hemiparese). Die Diagnostik erfolgt mit den gleichen Mittel wie im Erwachsenenalter: augenärztliche Untersuchung (Nachweis eines Papillenödem als Zeichen gesteigerten intrakraniellen Drucks) und vor allem eine Kernspintomographie des Schädels. Bei Kindern ist im Rahmen der multizentrischen Behandlungsstudien mittlerweile eine Kernspintomographie der gesamten Neuraxis (Gehirn und Rückenmark) erforderlich, um Abtropfmetastasen im Rückenmark nachzuweisen oder auszuschließen (spinale Aussaat). Ausgenommen von diesem Standard sind lediglich die Optikusgliome (Astrozytome des Sehnerven), wobei auch – extrem selten – Optikusgliome in das Rückenmark metastasieren können. Bei der Diagnostik sollte immer eine Lumbalpunktion erfolgen: diese dient zum Nachweis von Tumorzellen im Liquor (Nervenwasser) und Bestimmung von Tumormarkern im Liquor. Damit sollen andere Hirntumoren des Kindesalters (intrakranielle Keimzelltumoren, Medulloblastom, Ependymom) bereits präoperativ von einem Astrozytom abgegrenzt werden. Im Falle der Keimzelltumoren ist sogar ein operativer Eingriff bestenfalls zur Gewinnung einer histologischen Diagnose mittels Probebiopsie statthaft: diese Tumoren sprechen sehr gut auf Radio- und Chemotherapie an und sind daher auch primär mit diesen Methoden zu behandeln.
50 % der Operations-Resektate der radiologisch diagnostizierten hemisphärischen Astrozytome sind niedrig-maligne Astrozytome Grad I. Das Zystisch Juvenile Pilocystische Astrozytom ist eine gelegentliche Unterform. Der Rest sind Astrozytome Grad II, III und IV nach WHO. Bei Optikusgliomen (Astrozytom I Sehnerv) ist eine Operation infolge der drohenden schweren Schädigung des Sehvermögens zunächst nicht angezeigt. Die Behandlung erfolgt hier primär mit einer Kombination aus Chemotherapie und Strahlentherapie. Insbesondere bei jungen Kindern hat sich in den vergangenen Jahren die Protonentherapie etabliert,[17][18] da diese insbesondere für Kinder als besonders schonend gilt und somit langfristige Nebenwirkungen einer Strahlentherapie reduziert werden können.[19]
Die Behandlung erfolgt mit maximal totaler Resektion, soweit möglich. Patienten mit Astrozytom WHO Grad I und totaler Resektion erhalten nach der Operation radiologische Kontrollen in regelmäßigen zeitlichen Abständen, zumeist mittels Kernspintomographie. Astrozytom-Patienten mit signifikanten Residuen (Überresten des Tumors) nach der Operation werden bestrahlt. Patienten mit höhergradigen Astrozytomen (WHO Grad III und IV) erhalten Bestrahlung und Chemotherapie unabhängig vom Residualtumor. Astrozytome WHO I und II bei Kindern haben eine hohe 10-Jahre-Überlebensrate. Die Prognose eines Astrozytom WHO III (anaplastisches Astrozytom) ist deutlich schlechter, die Prognose eines Astrozytoms WHO IV (Glioblastom) sehr schlecht.
Einzelnachweise
- H. Yan, D. W. Parsons, et al.: IDH1 and IDH2 mutations in gliomas. In: The New England Journal of Medicine. Band 360, Nummer 8, Februar 2009, ISSN 1533-4406, S. 765–773, doi:10.1056/NEJMoa0808710, PMID 19228619, PMC 2820383 (freier Volltext). Seite 1, Results: „… mutations … of IDH1 in more than 70% of WHO grade II and III astrocytomas“, S. 5, Discussion: „… IDH1 mutations in 10 of 10 oligoastrocytomas and anaplastic oligoastrocytomas …“
- L. Dang et al.: Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. In: Nature. 2009; 462(7274), S. 739–744.
- C. L. Yu et al.: No association between residential exposure to petrochemicals and brain tumor risk. In: Cancer Epidemiol Biomarkers Prev. 2005; 14(12), S. 3007–3009.
- I. Deltour et al.: Time trends in brain tumor incidence rates in Denmark, Finland, Norway, and Sweden, 1974–2003. In: J Natl Cancer Inst. 2009; 101(24), S. 1721–1724.
- J. H. Scherer et al.: In: American Journal of Cancer. 1940; 40, S. 159–198. (no Pubmed entry)
- D. S. Russel, L. J. Rubinstein (Hrsg.): Pathology of Tumors of the Nervous System. Baltimore 1989.
- W. Muller et al.: In: Acta Neurochirurgica. 1977; 37, S. 75–91. PMID 195444
- E. R. Laws Jr. et al.: In: Journal of Neurosurgery. 1984; 61, S. 665–673. PMID 6470776
- J. M. Piepmeier et al.: In: Journal of Neurosurgery. 67: 177-181. PMID 3598677
- L. D. Recht et al.: In: Annals of Neurology. 1992; 31, S. 431–436. PMID 1586143
- J. Bouchard et al.: In: American Journal of Radiology. 1960; 84, S. 610–628. (no Pubmed Hit, perhaps: PMID 13472594)
- A. Gol: In: Journal of Neurosurgery. 1961; 18, S. 501–506. (no Pubmed Hit, perhaps: PMID 13655110)
- A. Uihlein et al.: Acta Radiologica. 1966; 5, S. 67–78. PMID 5954310
- D. M. Garcia et al.: In: Cancer. 1985; 55, S. 919–927. PMID 3967199
- L. Stephen: Hauser. In: Harrisons Neurologie. 17. Auflage. 2010, S. 502.
- Werner Hacke: Neurologie. 13. Auflage. Springer Verlag, 2010, S. 311.
- Protonentherapie bei Hirntumoren am WPE. In: WPE-UK.de. Westdeutsches Protonentherapiezentrum Essen (WPE), abgerufen am 5. August 2019.
- Medulloblastom (Kurzinformation). Abgerufen am 5. August 2019.
- B. Timmermann: Strahlentherapie bei Krebserkrankungen im Kindesalter – Wirkung, Chancen und mögliche Risiken. (PDF) Deutsche Kinderkrebsstiftung, 29. August 2018, abgerufen am 5. August 2019.
Nicht belegte Angaben beziehen sich im Wesentlichen auf:
- Andrew H. Kaye, Edward R. Laws Jr. (Hrsg.): Brain Tumors. Churchill Livingston, Edinburgh 1995, ISBN 0-443-04840-1.