Medulloblastom
Das Medulloblastom ist ein bösartiger embryonaler Tumor des Kleinhirns. Er tritt bevorzugt im Kleinkindes- und Kindesalter auf und ist in dieser Altersgruppe der häufigste bösartige Hirntumor. Er wird nach der WHO-Klassifikation der Tumoren des zentralen Nervensystems als Grad IV eingeordnet.
| Klassifikation nach ICD-10 | |
|---|---|
| C71 | Bösartige Neubildung des Gehirns |
| C71.6 | Zerebellum |
| ICD-10 online (WHO-Version 2019) | |
| Klassifikation nach ICD-O-3 | |
|---|---|
| 9470/3 | Medulloblastom o.n.A. Melanotisches Medulloblastom |
| 9471/3 | Desmoplastisches noduläres Medulloblastom Medulloblastom mit extensiver Nodularität |
| 9472/3 | Medullomyoblastom |
| ICD-O-3 erste Revision online | |
Epidemiologie
Jungen sind öfter betroffen als Mädchen, der Altergipfel liegt je nach Literatur zwischen 1–9 Jahren oder 4–8 Jahren. Rund ein Fünftel der Medulloblastome treten allerdings auch nach der Pubertät auf, dann mit einem Altersgipfel im dritten Lebensjahrzehnt.
Symptome
Medulloblastome infiltrieren beide Kleinhirnhemisphären und können ebenfalls in den Liquorraum metastasieren ("Abtropfmetastasen"). Klinisch stehen meist Störungen der Kleinhirnfunktion (zum Beispiel Fallneigung und Ataxie) sowie Zeichen des erhöhten Hirndruckes mit Kopfschmerzen, Lethargie und morgendlichem Erbrechen im Vordergrund. Der erhöhte Hirndruck entsteht einerseits durch die tumorgewebebedingte intrakranielle Volumenerhöhung selbst, andererseits durch den sich schnell entwickelnden Verschlusshydrocephalus. Bei Spiegelung des Augenhintergrundes kann eine Stauungspapille als Zeichen für erhöhten Hirndruck erkannt werden. Die Einklemmung von Kleinhirnanteilen und Kompression der Medulla oblongata (die ihrerseits Sitz des lebensnotwendigen Atem- und Kreislaufzentrums ist) bedingten oft den Tod der jungen Patienten.
Bildgebung
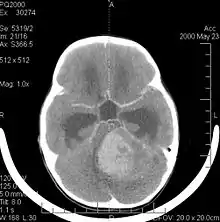
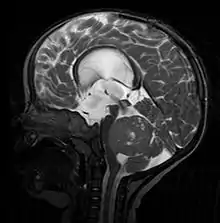
In der Computertomographie- und Magnetresonanztomographie (MRT) stellen sich Medulloblastome als solide Raumforderungen mit inhomogener Kontrastmittelaufnahme dar. Sie wachsen meist vom Boden des 4. Ventrikels in den Kleinhirnwurm. Im nativen CT ist der Tumor gering hypodens, nach KM-Gabe hyperdens. Im MRT kann man den Tumor aufgrund seiner inhomogenen, symmetrisch in der Mittellinie gelegenen Lage in kraniokaudaler Ausdehnung mit hypointensem Signal in T1 und hyperintensem Signal in T2 (weil vermehrt wasserreich) gut erkennen. Oft kommt es zu einer Obstruktion der Ventrikel; als Folge dessen kommt es zum Hydrocephalus. Bei sehr jungen Patienten kann sich dies durch verbreiterte Kalottennähte (Wolkenschädel) zeigen, welche als Konsequenz des steigenden Hirndrucks auseinanderweichen.
Pathologie
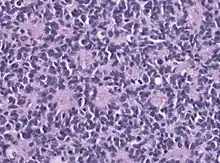
Das Medulloblastom ist ein undifferenzierter embryonaler Tumor, der von embryonalen pluripotenten Zellen abstammt. Feingeweblich sind Medulloblastome zelldichte klein- rund- und blauzellige Tumoren. Die Tumorzellen weisen runde bis ovale Kerne auf und können in Rosettenformationen angeordnet sein. Immunhistochemisch ist eine Expression neuronaler Marker (Synaptophysin, Neurofilament, NeuN) typisch aber auch gliale Proteine wie GFAP können kleinflächig exprimiert werden. Als Varianten abzugrenzen sind desmoplastisches Medulloblastom, anaplastisches Medulloblastom und Medulloblastom mit extensiver Nodularität, Desmoplastisches noduläres Medulloblastom, Medullomyoblastom
Genetik
Der erste Nachweis einer p53-Mutation in einem Medulloblastom erfolgte in den frühen Neunzigern.[1] Derzeit werden Medulloblastome anhand ihres genetischen Profils in die vier folgenden Subgruppen eingeteilt:[2]
- WNT: meist Kinder, selten Erwachsene: Monosomie Chromosom 6, CTNNB1-Mutationen, meist klassische Medulloblastome, selten großzellige/anaplastische Medulloblastome. Diese Gruppe hat die beste Prognose.
- SHH (Sonic hedgehog): Kleinkinder (meist desmoplastisch/nodulär) und Erwachsene (klassisch oder großzellig/anaplastisch): PTCH/SMO/SUFU Mutationen, GLI2 Amplifikationen.
- Gruppe 3: meist Kinder, klassische Medulloblastome, häufig metastasierend, MYC-Amplifikation, Chromosomale Verluste auf 5q und 10q. Diese Gruppe hat die schlechteste Prognose.
- Gruppe 4: meist Kinder, häufiger männlich, häufig Isochromosom 17q. Chromosomale Verluste auf 8 und X.
Therapie
Aktuelle Behandlungskonzepte für Kinder und Jugendliche mit einem Medulloblastom sehen eine neurochirurgische Tumorentfernung, eine Chemotherapie und eine Strahlentherapie vor. Statt der herkömmlichen Strahlentherapie kann auch eine Protonentherapie erfolgen[3], die noch zielgerichteter und schonender wirkt und daher eine immer größere Bedeutung bei der Behandlung von Tumoren im Kindes- und Jugendalter gewinnt.[4][5] Sofern möglich, sollte immer eine komplette Tumorresektion angestrebt werden. Oft ist dies aufgrund der Lage oder der Infiltration lebenswichtiger Strukturen nur begrenzt möglich, sodass nur teilreseziert werden kann. Dies kann allerdings auch zur Wiederherstellung der Liquorpassage dienen.
Prognose
Der Krankheitsverlauf hängt unter anderem von der Tumorgröße, dem Ausmaß der Tumorentfernung, einer etwaigen Absiedlung von Tumorzellen entlang der Liquorwege und dem Alter des Patienten ab. Auch die Art der feingeweblichen Differenzierung kann eine prognostische Rolle spielen, wobei bei Patienten mit einem sogenannten desmoplastischen Medulloblastom der Krankheitsverlauf günstiger ist.
Die Patienten werden in klinischen Studien in Niedrig-, Standard- und Hochrisikogruppen eingeteilt:
- Bei Patienten der Niedrigrisikogruppe (in der Regel WNT-aktiviert) werden je nach Studie Heilungsraten von bis zu 100 % erreicht.[6] Die derzeitigen Bemühungen bewegen sich deshalb in die Richtung, die Intensität der Therapie und damit die negativen Langzeitfolgen zu reduzieren aber gleichzeitig die hohen Heilungsraten zu bestätigen.[7]
- In der HIT-SIOP PNET 4 Studie, an welcher 340 Kinder und Jugendliche der Standardrisikogruppe im Alter von vier bis 21 Jahren aus mehreren europäischen Ländern teilnahmen, betrug die 5-Jahres-Überlebensrate je nach Randomisierung zwischen 85 % und 87 %. Rund 78 % der Patienten blieben während 5 Jahren ohne Rückfall und gelten damit als geheilt.[8] Nach einem Rückfall war die Prognose sehr schlecht. Von 66 Patienten waren 5 Jahre nach einem Rückfall trotz intensiver Behandlung nur noch vier am Leben.[9]
- An einer US-amerikanischen Studie nahmen 161 Patienten im Alter zwischen drei und 21 Jahren mit einem Hochrisiko-Profil teil. Je nach Randomisierung erhielt die Hälfte der Patienten während der Bestrahlung zusätzlich täglich Carboplatin. Die 5-Jahres-Überlebensrate der Patienten mit Carboplatin betrug 82 %, derjenigen ohne 68 %.[10] Derzeit und bis April 2024 findet die europäische SIOP PNET 5 Studie statt, bei welcher man die vielversprechenden Ergebnisse mit Carboplatin während der Bestrahlung bei der Standardrisikogruppe zu bestätigen versucht.[11]
Bei Neugeborenen und Kleinkindern unter drei Jahren wird aufgrund der enormen Nebenwirkungen in der Regel auf eine Bestrahlung verzichtet oder mithilfe von Chemotherapien so lange hinausgeschoben, bis die Patienten das vierte Lebensjahr erreichen. In einer klinischen Studie, an der 62 Patienten teilnahmen, betrug die 5-Jahres-Überlebensrate 66 % bei Kindern, die nach Diagnose lediglich intensiv chemotherapiert worden waren. Nach vollständiger Tumorentfernung konnte hier eine 5-Jahres-Überlebensrate von 93 % erreicht werden. Nach unvollständiger Tumorentfernung oder Absiedlung von Tumorzellen entlang der Liquorwege betrugen die 5-Jahres-Überlebensraten 56 % bzw. 38 %.[12]
Literatur
- D. D. Louis, H. Ohgaki, O. Wiestler u. a. (Hrsg.): World Health Organization classification of tumors. Pathology and genetics of tumours of the nervous system. IARC Press, Lyon 2007, ISBN 978-92-832-2430-3.
- S1-Leitlinie Medulloblastom im Kindes- und Jugendalter der Deutschen Gesellschaft für Kinder- und Jugendmedizin (DGKJ). In: AWMF online (Stand 2008)
Weblinks
Einzelnachweise
- H. Ohgaki, R. H. Eibl, O. D. Wiestler, M. G. Yasargil, E. W. Newcomb, P. Kleihues: p53 mutations in nonastrocytic human brain tumors. In: Cancer Research. 1991 Nov 15;51(22), S. 6202–6205. PMID 1933879 Volltext
- M. D. Taylor, P. A. Northcott, A. Korshunov, M. Remke, Y. J. Cho, S. C. Clifford, C. G. Eberhart, D. W. Parsons, S. Rutkowski, A. Gajjar, D. W. Ellison, P. Lichter, R. J. Gilbertson, S. L. Pomeroy, M. Kool, S. M. Pfister: Molecular subgroups of medulloblastoma: the current consensus. In: Acta neuropathologica. Band 123, Nummer 4, April 2012, S. 465–472, ISSN 1432-0533. doi:10.1007/s00401-011-0922-z. PMID 22134537. PMC 3306779 (freier Volltext).
- Protonentherapie bei Hirntumoren am WPE | WPE-UK.de. In: Westdeutsches Protonentherapiezentrum Essen (WPE). Abgerufen am 5. August 2019 (deutsch).
- Medulloblastom (Kurzinformation). Abgerufen am 5. August 2019.
- B. Timmermann: Strahlentherapie bei Krebserkrankungen im Kindesalter. (PDF) In: https://www.kinderkrebsstiftung.de/. Deutsche Kinderkrebsstiftung, 29. August 2018, abgerufen am 5. August 2019.
- Identifying Low-Risk Medulloblastoma to De-escalate Therapy. Abgerufen am 29. Dezember 2019.
- International Society of Paediatric Oncology (SIOP) PNET 5 Medulloblastoma - Full Text View - ClinicalTrials.gov. Abgerufen am 29. Dezember 2019 (englisch).
- Birgitta Lannering, Stefan Rutkowski, Francois Doz, Barry Pizer, Göran Gustafsson: Hyperfractionated Versus Conventional Radiotherapy Followed by Chemotherapy in Standard-Risk Medulloblastoma: Results From the Randomized Multicenter HIT-SIOP PNET 4 Trial. In: Journal of Clinical Oncology. Band 30, Nr. 26, 30. Juli 2012, ISSN 0732-183X, S. 3187–3193, doi:10.1200/JCO.2011.39.8719 (ascopubs.org [abgerufen am 29. Dezember 2019]).
- Magnus Sabel, Gudrun Fleischhack, Stephan Tippelt, Göran Gustafsson, François Doz: Relapse patterns and outcome after relapse in standard risk medulloblastoma: a report from the HIT-SIOP-PNET4 study. In: Journal of Neuro-Oncology. Band 129, Nr. 3, September 2016, ISSN 1573-7373, S. 515–524, doi:10.1007/s11060-016-2202-1, PMID 27423645, PMC 5020107 (freier Volltext).
- Regina I. Jakacki, Peter C. Burger, Tianni Zhou, Emiko J. Holmes, Mehmet Kocak: Outcome of Children With Metastatic Medulloblastoma Treated With Carboplatin During Craniospinal Radiotherapy: A Children's Oncology Group Phase I/II Study. In: Journal of Clinical Oncology. Band 30, Nr. 21, 20. Juli 2012, ISSN 0732-183X, S. 2648–2653, doi:10.1200/JCO.2011.40.2792, PMID 22665539, PMC 4559602 (freier Volltext).
- International Society of Paediatric Oncology (SIOP) PNET 5 Medulloblastoma - Full Text View - ClinicalTrials.gov. Abgerufen am 29. Dezember 2019 (englisch).
- Rutkowski et al.: Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. In: N Engl J Med. 2005; 352, S. 978–86. PMID 15758008 Volltext