Pituizytom
Das Pituizytom ist ein seltener, gutartiger (WHO Grad I), von der Hypophyse ausgehender Hirntumor, der meist im Erwachsenenalter auftritt.[1] Der Tumor entsteht aus den nur in der Neurohypophyse vorkommenden, auf Transport, Speicherung und Freigabe der Hormone Antidiuretisches Hormon und Oxytocin spezialisierten, Pituizyten genannten Gliazellen.[2][3]
| Klassifikation nach ICD-10 | |
|---|---|
| C75.1 | Bösartige Neubildung sonstiger endokriner Drüsen und verwandter Strukturen – Hypophyse |
| C71.9 | Bösartige Neubildung des Gehirns – Gehirn, nicht näher bezeichnet |
| ICD-10 online (WHO-Version 2019) | |
| Klassifikation nach ICD-O-3 | |
|---|---|
| 9432/1 | Pituizytom |
| ICD-O-3 erste Revision online | |
Vor 2016 wurde der Begriff „Pituizytom“ breiter gefasst und schloss Granularzelltumor in der Hypophyse und Pilozytische Astrozytome der Neurohypophyse mit ein.[3]
Pathologie
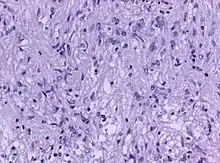
Die Tumoren sind gut abgegrenzt, eher weich bis mittelfest und haben eine graue bis gelbe gleichmäßige oder körnige Oberfläche. Nekrosen und Zystenbildungen sind selten. Histologisch bestehen sie aus Spindelzellen und Sternzellen (Stellate cells) und sind gut durchblutet.[3]
Verbreitung
Die Häufigkeit wird mit unter 0,4 % aller Tumoren der Hyophysenregion angegeben.[4] Meist treten Pituizytome zwischen dem 4. und dem 6. Lebensjahrzehnt auf, das männliche Geschlecht ist etwas häufiger betroffen.[3]
Klinische Erscheinungen
Klinische Kriterien sind:[2][1][3]
- Lokalisation intrasellär (in der Hypophysengrube) oder suprasellär
- typische Hinweise sind Sehstörungen, Kopfschmerz und Hypophyseninsuffizienz
Diagnose
Die Diagnose stützt sich auf Bildgebung durch Magnetresonanztomographie oder Computertomographie und wird durch Biopsie gesichert.[3]
Differentialdiagnostik
Abzugrenzen sind:[3]
- Granularzelltumor der Hypophysenregion
- Pilozytisches Astrozytom der Neurohypophyse
- Kraniopharyngeom
- Hypophysenadenom (Makroadenom)
- Meningeom
- Metastase und Tumorinfiltration der Hypophyse bei lymphozytischer Hypophysitis oder Neurosarkoidose
- Optikusgliom der Sehbahn
Therapie
Die Behandlung besteht in – soweit möglich – vollständiger Resektion.[1][5] Solange keine raumfordernde Wirkung besteht, kann abgewartet und beobachtet werden, da der Tumor gutartig ist und nur langsam wächst. Aufgrund der ungünstigen anatomischen Lage kann es zu Rezidiven kommen.[3] Die Prognose gilt als gut.[2]
Literatur
- X. Li, Y. Liu, Y. Miao, J. Wang, L. Wang, E. H. Wang: A rare case of pituicytoma presenting with severe Cushing disease: A case report and review of literature. In: Medicine. Band 98, Nummer 44, November 2019, S. e17772, doi:10.1097/MD.0000000000017772, PMID 31689841, PMC 6946303 (freier Volltext) (Review).
- J. A. Ellis, N. M. Tsankova, R. D'Amico, J. C. Ausiello, P. Canoll, M. K. Rosenblum, J. N. Bruce: Epithelioid pituicytoma. In: World Neurosurgery. Band 78, Nummer 1–2, Juli 2012, S. 191.E1–191.E7, doi:10.1016/j.wneu.2011.09.011, PMID 22120271, PMC 3926508 (freier Volltext) (Review).
Einzelnachweise
- Pschyrembel online
- Pituizytom. In: Orphanet (Datenbank für seltene Krankheiten).
- Radiopaedia.org
- AWMF S2k-Leitlinie: Diagnostik und Therapie klinisch hormoninaktiver Hypophysentumoren, April 2020, AWMF
- M. Feng, J. D. Carmichael, V. Bonert, S. Bannykh, A. N. Mamelak: Surgical management of pituicytomas: case series and comprehensive literature review. In: Pituitary. Band 17, Nummer 5, Oktober 2014, S. 399–413, doi:10.1007/s11102-013-0515-z, PMID 24037647 (Review).