Lhermitte-Duclos-Syndrom
Das Lhermitte-Duclos-Syndrom (LDD, auch Dysplastisches Gangliozytom) ist eine seltene Neubildung des Kleinhirns aufgrund einer Mutation im PTEN-Gen und gehört daher zum PTEN-Hamartoma-Tumor-Syndrom. Es wird auch als Unterform des Cowden-Syndroms beschrieben.
| Klassifikation nach ICD-11 | |
|---|---|
| XH6K00 | Dysplastic gangliocytoma of cerebellum (Lhermitte-Duclos) |
| ICD-11 (WHO-Version 2019) | |
Die Erstbeschreibung des Leidens erfolgte 1920 durch Jacques Jean Lhermitte und P. Duclos.
Pathologie und Histopathologie
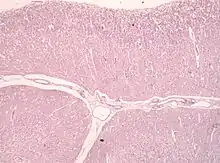
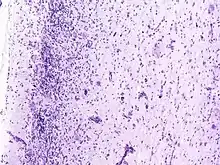
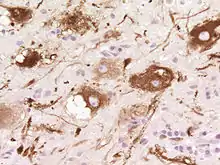
Nach der WHO-Klassifikation der Tumoren des zentralen Nervensystems zählt es zu den Grad I Tumoren. Das Syndrom zeigt allerdings gleichzeitig Charakteristika einer gutartigen Neubildung und einer Fehlbildung, so dass die genaue Einordnung noch unklar ist. Auch ob es sich um ein Hamartom handeln könnte, ist in Fachkreisen umstritten.
Bei der Entstehung spielt das Tumorsuppressorgen PTEN eine Rolle. Dem Cowdensyndom liegt eine Keimbahnmutation des PTEN-Gens zugrunde. In Mausmodellen konnte die Rolle des PTEN-Gens bei der Pathogenese des Lhermitte-Duclos-Syndroms nachgewiesen werden: Eine gezielte Ausschaltung von PTEN im Kleinhirn führt zu einer Faltungs- und Zellwanderungsstörung ähnlich der Architekturstörungen beim Menschen.
Auf zellulärer Ebene sieht man dysplastische (hypertrophe und später blasig aufgetriebene, vakuolisierte) Purkinjezellen. Die Schichtung des Kleinhirns ist umgekehrt (invertiert) und die Körnerzellschicht ist weitgehend aufgelöst.
Symptome, Diagnostik und Therapie
Zu den klinischen Symptomen und Zeichen gehören Kopfschmerzen, Bewegungsstörungen (wie z. B. ein Tremor), Sehstörungen und eine Vergrößerung des Kopfes, die einen Hydrozephalus imitieren kann. Die Beschwerden nehmen über Monate bis Jahre langsam zu.
Das Elektroenzephalogramm kann verändert sein. Die Magnetresonanztomographie bietet heute eine sichere Nachweismethode, beweisend ist der histopathologische Befund.
Die Therapie besteht in der operativen Entfernung der Tumormasse. Rezidive sind allerdings häufig. In einigen Fällen mit familiärer Häufung konnte eine autosomal-dominante Vererbung des Leidens bewiesen werden.
Literatur
- J. Lhermitte, P. Duclos: Sur un ganglioneurome diffuse du cortex du cervelet. Bulletin de l’Association francaise pour l'etude du cancer, Paris, 1920, 9: 99–107. (Erstbeschreibung)
- R. Kumar, V. K. Vaid, S. K. Kalra: Lhermitte-Duclos disease. In: Child’s nervous system. Band 23, Nummer 7, Juli 2007, S. 729–732, ISSN 0256-7040. doi:10.1007/s00381-006-0271-8. PMID 17221273.
- N. Vantomme, F. Van Calenbergh, J. Goffin, R. Sciot, P. Demaerel, C. Plets: Lhermitte-Duclos disease is a clinical manifestation of Cowden’s syndrome. In: Surgical Neurology. Band 56, Nummer 3, September 2001, S. 201–204, ISSN 0090-3019. PMID 11597654. (Review).
- T. Boonpipattanapong, N. Phuenpathom, W. Mitarnun: Cowden’s syndrome with Lhermitte-Duclos disease. In: British journal of neurosurgery. Band 19, Nummer 4, August 2005, S. 361–365, ISSN 0268-8697. doi:10.1080/02688690500305431. PMID 16455548. (Review).