Chordom
Chordome sind langsam und destruktiv wachsende Tumoren der Wirbelsäule und der Schädelbasis, die in etwa 10 % der Fälle metastasieren.
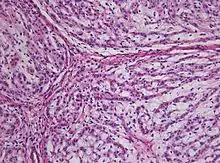
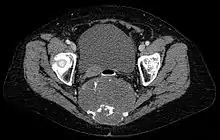
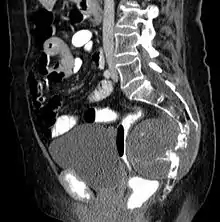
Chordome werden manchmal zu den Knochentumoren gezählt, obwohl sie nicht aus Knochengewebe stammen, sondern aus Resten der Chorda dorsalis (Notochordoa) an den Enden der Wirbelsäule. Damit lassen sich auch ihre Hauptlokalisationen, nämlich Schädelbasis und Steißbein, erklären. Die ICD-O-3 klassifiziert sie unter die Sonstigen Tumoren des Nervensystems (Nr. 937).
Epidemiologie
Chordome machen etwa 1 % aller Knochentumoren aus und treten in der Regel erst jenseits des 30. Lebensjahr auf. Männer und Frauen sind ungefähr gleichhäufig betroffen.[1]
Symptome
Symptome entstehen besonders durch Druck auf Nerven durch den Tumor, was allgemein zu Schmerzen und Nervenausfällen führen kann. Chordome im Bereich der Schädelbasis führen so zu Kopf- und Nackenschmerzen, Diplopie und weiteren Hirnnervenausfällen[1].
Therapie
Die Therapie besteht in der chirurgischen Entfernung. Da diese nur in 30 % der Fälle vollständig gelingt, folgt in der Regel eine Strahlentherapie, um die Rezidiv-Rate weiter zu verringern[1]. Trotzdem kommt es in zwei Drittel der Fälle zu einem Lokalrezidiv des Tumors[1]. Chordome sind chemotherapieresistent[1]. Studien legen eine Wirksamkeit einer adjuvanten Therapie mit Imatinib und Lapatinib[2] nahe.
Gen-Analysen an elf Patienten, denen keine Standardtherapie mehr half, ergaben bei drei Patienten eine gestörte DNA-Reparatur. Ein bei anderen Krebsarten mit mangelnder homologer Rekombination zugelassener PARP-Inhibitor stoppte in einer experimentellen Behandlung das Tumorwachstum zehn Monate – bis eine neue Resistenzmutation die Wirkung der Arznei aufhob.[3]
Prognose
Die 5-Jahres-Überlebensrate beträgt 63–78 %, die 10-Jahres-Überlebensrate 16–32 %.
Literatur
- Heuck, Wörtler, Vestring: Radiologie der Knochen- und Gelenkerkrankungen: Primäre und sekundäre Knochentumoren. Thieme Verlag, 1997, ISBN 3-13-107071-4
Einzelnachweise
- Jason K. Wasserman, Denis Gravel, Bibianna Purgina: Chordoma of the Head and Neck: A Review. In: Head and Neck Pathology. 4. Oktober 2017, ISSN 1936-0568, doi:10.1007/s12105-017-0860-8, PMID 28980142.
- Phase II study on lapatinib in advanced EGFR-positive chordoma, Annals of Oncology 00: 1–6, 2013, doi:10.1093/annonc/mdt117.
- Stefan Gröschel, Daniel Hübschmann, Francesco Raimondi, et al. (2019) Defective homologous recombination DNA repair as therapeutic target in advanced chordoma. Nature Communications, https://doi.org/10.1038/s41467-019-09633-9