Wittig-Reaktion
Die Wittig-Reaktion ist eine organisch-chemische Reaktion, die der Knüpfung von C=C-Bindungen dient und nach ihrem Entdecker Georg Wittig benannt ist. Mit ihr lassen sich Carbonylverbindungen (Aldehyde oder Ketone) mit Phosphoryliden unter Substitution des Carbonylsauerstoffs zu Alkenen olefinieren:

Als Olefinbildungsreaktion hat sie in Labor und Industrie große Bedeutung. Die Wittig-Reaktion ist eine breit anwendbare Methode zur Synthese von Olefinen und toleriert dabei vielfältige funktionelle Gruppen. Als störend für die Wittig-Reaktion erweisen sich normalerweise nur recht saure funktionelle Gruppen wie Carbonsäuren oder 1,3-Dicarbonylverbindungen.
Allgemeines
Bei der Wittig-Reaktion sind sowohl inter- als auch intramolekulare Varianten bekannt. Die Reaktion ist regioselektiv, das heißt, die neu gebildete Kohlenstoff-Kohlenstoff-Doppelbindung findet sich an der Stelle der früheren Kohlenstoff-Sauerstoff-Doppelbindungen wieder. Als Carbonyl-Komponente kommen Aldehyde und Ketone in Frage, wobei Aldehyde reaktiver sind und sterisch weniger gehinderte Ketone gegenüber sterisch gehinderten Ketonen selektiv olefiniert werden können. Carbonsäureester sind in einer Wittig-Reaktion nahezu inert.
Ein Nachteil ist die prinzipiell wenig ausgeprägte (E,Z)-Selektivität, die aber durch geeignete Reaktionsbedingungen gesteuert werden kann. Durch seine hohe Toleranz an funktionellen Gruppen kann die Wittig-Reaktion auch im Wittig-Reagenz eine Vielzahl an funktionellen Gruppen selber mitbringen.
Einsatz findet die Wittig-Reaktion bei der Synthese olefinischer Naturstoffe wie Vitamin A und D, Carotinoiden (β-Carotin s. u.), Squalen, ungesättigten Pheromonen, Insektenhormonen, Riechstoffen und Prostaglandinen.
Geschichte
Entdeckung
Wittig hat bei seinen Experimenten zur Quaternisierung der Hauptgruppenelemente ein Zwitterion erhalten, welches bemerkenswerte Eigenschaften aufwies.
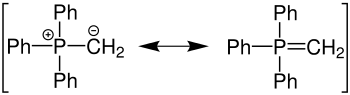
In Gegenwart von Carbonyl-Verbindungen reagierten sie glatt mit dem Carbonyl-Kohlenstoff unter Ausbildung einer Kohlenstoff-Kohlenstoff-Doppelbindung und Triphenylphosphinoxid.[1][2]
Wittig publizierte diese 1947 an der Universität Tübingen entdeckte neuartige Reaktion unter dem Titel Über Triphenyl-phosphin-methylene als olefinbildende Reagenzien und nannte diese Reaktion fortan Carbonyl-Olefinierung. Der Name Wittig-Reaktion wurde später jedoch gebräuchlich. Heute wird in der Regel das Triphenylalkylphosphoniumsalz als Wittig-Salz für diese Reaktion eingesetzt und man erhält als Nebenprodukt Triphenylphosphinoxid.
Die Wittig-Reaktion erwies sich schnell als sehr vielseitig und universell einsetzbar. Georg Wittig wurde 1979 mit dem Nobelpreis für Chemie ausgezeichnet.
Erste industrielle Anwendungen
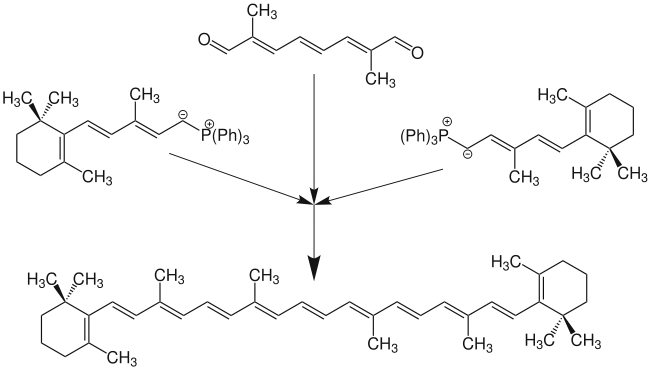
Mit Begeisterung nahm die BASF die neue Methode zur Kenntnis. Sie erlaubte es, einen bereits vorhandenen Synthesebaustein [Derivat von (2E,4E)-3-methyl-5-(2,6,6-trimethylcyclohex-1-en-1-yl)pentan] mit dem leicht zugänglichen (2E,4E,6E)-2,7-dimethylocta-2,4,6-trienedial zu β-Carotin zu verknüpfen. Bereits drei Jahre nach der Publikation von Wittig wurde ein Patent eingereicht (BASF).[3][4]
Herstellung der Ylide
Trisubstituierte Phosphoralkyle/aryle lassen sich in einer SN2-Reaktion quaternisieren. Triphenylphosphin (1) reagiert beispielsweise mit Ethylbromid (2) unter Bildung von Phosphoniumsalz über Ethyltriphenylphosphoniumbromid (3) zu einem Ylid (4) und einem Ylen:

Triphenylphosphin (1) und Ethylbromid (2) werden im Autoklaven in Benzol 20 h auf 130 °C erhitzt. Bei Abkühlen fällt das Phosphoniumsalz kristallin aus (Ausbeute 90 %).[5]
Phosphine sind gute Nucleophile, aber schlechte Basen. Aus diesem Grund wird die der SN2-Reaktion konkurrierende E2-Eliminierung fast vollständig unterdrückt. So lassen sich die meisten primären und sekundären Alkylhalogenide in guten Ausbeuten in ihre Phosphoniumsalze überführen.
Im Gegensatz zu den meist isolierten Phosphoniumhalogeniden werden die daraus durch eine Deprotonierung am α-C-Atom hergestellten Ylide meist direkt weiter umgesetzt.
Unter Inertgas in DMSO wird das Ethyltriphenylphosponiumbromid (3) mit Natriumhydrid deprotoniert.[5]
Es wird eine nach außen neutrale Phosphorverbindung gebildet, die man als Ylid (4) oder Phosphoran bezeichnet. Diese Betaine lassen sich aber auch als Ylen formulieren. Andere gängige Deprotonierungsreagenzien sind Phenyllithium oder n-Butyllithium.
Eine moderne und einfache Methode zur Deprotonierung des Phosphoniumions unter lithiumfreien Bedingungen ist die Verwendung von Kalium-tert-butanolat in THF oder bei sterisch gehinderten Carbonylgruppen in Toluol. Dabei kann das Wittig-Salz trocken in äquimolaren Verhältnissen mit der Base vermengt und mit dem Lösungsmittel versetzt werden, was eine einfacher zu handhabende Alternative zu dem Instant-Ylid darstellt.[6][7][8] Phosphor-Ylide lassen sich auch durch Carbenaddition an Phosphine erhalten.[9]
Mechanismen der Wittig-Reaktion
Der Mechanismus der Wittig-Reaktion kann allgemein oder aus stereochemischer Sicht betrachtet werden.
Wittig-Reaktion, allgemein
Der folgende Mechanismus erklärt die allgemeine Wittig-Reaktion, bei der Phosphorylide mit Carbonylverbindungen zu Alkenen reagieren.
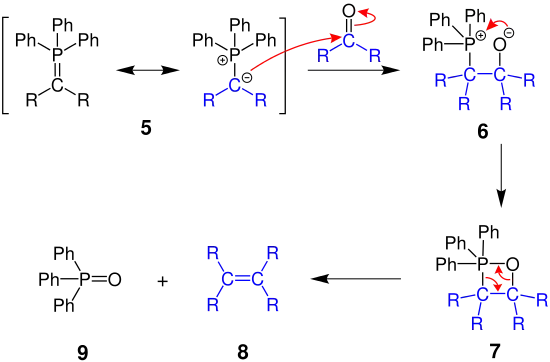
Das negativ geladene Kohlenstoffatom des Phosphorylids 5 greift die Carbonylverbindung des Edukts an und es entsteht ein Phosphor-Betain 6, welches weiter zum Oxaphosphetan 7, einem viergliedrigen Ring, reagiert. Dieser zerfällt abschließend in das Alken 8 und Triphenylphosphanoxid 9, aufgrund der starken Phosphor-Sauerstoff-Doppelbindung.[10]
Wittig-Reaktion, stereochemische Aspekte
Insgesamt ist der Mechanismus noch nicht genau geklärt, bzw. er läuft je nach Substituenten verschieden ab. Zum Großteil läuft die Reaktion mehrstufig ab; einzelne Zwischenverbindungen lassen sich isolieren. Bei reaktiven Yliden/Carbonyl-Verbindungen erfolgt der Angriff des Carbanions und Bildung des Oxaphosphetans konzertiert nach den Woodward-Hoffmann-Regeln.[11] Es gibt ebenfalls Hinweise, dass in Gegenwart von sehr großen Substituenten (hohe sterische Hinderung), die Reaktion über radikalische Stufen (SET single electron transfer) verläuft.[12]
Bereits bei der Bildung des Betains wird festgelegt, ob das Alken (E)- oder (Z)-konfiguriert ist. Der erste Schritt ist jedoch teilweise reversibel.
- Bei reversibler Addition wird in diesen Fällen das thermodynamisch stabilere Additionsprodukt gebildet (thermodynamische Reaktionskontrolle).
- Bei irreversibler Addition wird das Produkt gebildet, das sich schneller bildet (niedrigere Aktivierungsenergie; kinetische Reaktionskontrolle).[13]
Schon kurz nach der Entdeckung erkannte man, dass die Wittig-Reaktion meist recht diastereoselektiv verläuft. Je nach Reaktivität der eingesetzten Ylid/Carbonyl-Komponenten kann die Diastereoselektivität durch Wahl der Substituenten (sowohl am Ylid als auch an der Carbonylverbindung) und den Reaktionsbedingungen das Ausbeuteverhältnis der Isomere gezielt verändert werden.[14] Da die Reaktivität des Carbonyls in der Regel gegeben ist, versucht man die Diastereoselektivität durch die Anpassung der Reaktivität des Ylids zu beeinflussen. Generell gilt:
| Reaktivität der Carbonylverbindung | ||||
|---|---|---|---|---|
| hoch | mittel | niedrig | ||
| Reaktivität des Ylides | ||||
| hoch | (Z) | (Z) | (E,Z) | |
| mittel | (Z) | (E,Z) | (E,Z) | |
| niedrig | (E) | (E) | (E) | |
Möglichkeiten der Variation der Reaktivität des Oxaphosphetan
Die Destabilisierung kann durch Ersatz der Substituenten am Phosphor mit π/σ-Donatoren erfolgen. Es entstehen dann meist mit hohen Ausbeuten die (Z)-Olefine.
Die Stabilisierung kann durch Ersatz der Substituenten am Phosphor mit π/σ-Akzeptoren erfolgen. Es entstehen dann meist mit hohen Ausbeuten die (E)-Olefine.
Mehrstufig (weniger reaktive Komponenten)
Eingeleitet wird die Reaktion durch den Angriff des Carbanions an das positiv polarisierte Carbonyl-Kohlenstoff-Atom und der Bildung des Betains. Die Carbonyl-Verbindung ist in den meisten Fällen prochiral, d. h. bei der Betain-Bildung wird ein Chiralitätszentrum neu gebildet, das je nach Angriffsseite (pro-R, bzw. pro-S) (R)- oder (S)-konfiguriert ist.
Das Carbanion des Ylides ist ebenfalls prochiral. Es kommt also intermediär zur Bildung einer Verbindung, die zwei benachbarte chirale C-Atome enthält. Je nach Art der Anordnung der Substituenten kommt es zu zwei unterschiedlichen Formen, die sich auf die Kohlenhydrate Threose und Erythrose zurückführen lassen. Zur Benennung ordnet man die Substituenten nach Größe. Ergibt sich eine Konformation (durch Rotation um die C-C-Achse), in der sich die großen, mittleren und kleinen Substituenten jeweils gegenüberstehen, spricht man von der erythro-Form, alternativ von der threo-Form. Das Durchlaufen der Betain-Zwischenstufe konnte durch Isolierung stabiler Vertreter nachgewiesen werden.[15]
Das Betain reagiert über das Konformere, das eine thermische [2+2]-Cycloaddition zum cyclischen Oxaphosphetan ermöglicht. Die Existenz des Vierring-Intermediates konnte durch 31P-NMR-Spektroskopie nachgewiesen werden.[16][17] Das bei −80 °C gebildete Oxaphosphetan ist bei diesen Temperaturen stabil.[18] Beim Erwärmen auf 0 °C zersetzt es sich.
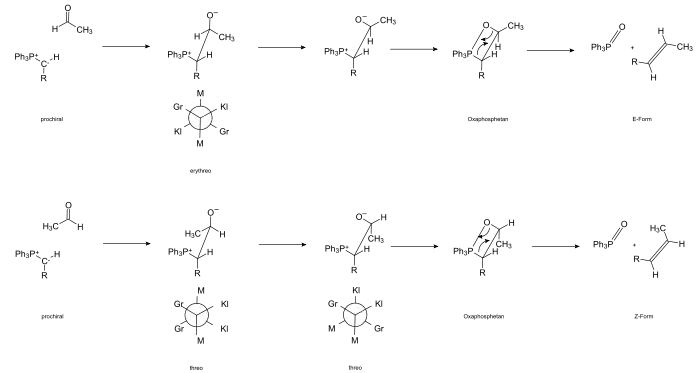
Durch eine retro-[2+2]-Cycloaddition kommt es zur Ausbildung von Triphenylphosphinoxid und dem (E)-Alken.
Konzertiert (reaktivere Komponenten)
Bei nicht stabilisierten Yliden verläuft die Reaktion schnell. Durch Coulomb-Anziehung nähern sich die Reaktanten orthogonal (jeweils die positiv polarisierten Atome lagern sich an jeweils negativ geladenen an). Gleichzeitig positionieren sich die „großen“ Methylgruppen aus sterischen Gründen möglichst weit voneinander weg. Die Bindung bildet sich durch Torsion des Übergangkomplexes und konzertierter [2s+2a]-Cycloaddition (disrotatorischer Ringschluss).[19] Die Betain-Zwischenstufe wird quasi übersprungen.
Über einen „verdrillten“ Vierring (twisted) bildet sich schließlich das Z-Oxaphosphetan.
Nebenreaktionen
Nicht stabilisierte Ylide sind labil gegenüber Sauerstoff und Wasser. Durch Hydrolyse entsteht aus einem Ylid ein Phosphinoxid und ein Kohlenwasserstoff.
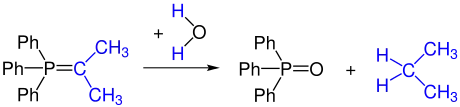
Durch Teilumsetzung mit Sauerstoff (oder durch Zugabe von Oxidationsmitteln) kann man einen Teil des Ylids zur Carbonylverbindung reoxidieren. Diese Carbonylverbindung reagiert mit einem weiteren Ylid zu einem Alken.
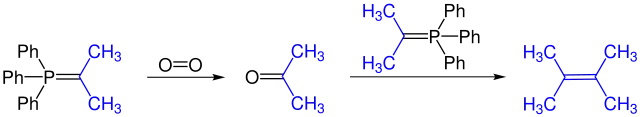
Gewöhnlich sind diese Nebenreaktionen unerwünscht und ausbeutevermindernd, weshalb man unter Inertgas und Feuchtigkeitsausschluss arbeitet. Teilweise werden diese Reaktionen aber auch präparativ genutzt.[20]
Enantioselektive Wittig-Reaktionen
Durch Verwendung chiraler Phosphorliganden erzielten Trost und Curran[21] in einer intramolekularen Wittigreaktion einen Enantiomerenüberschuss (ee) von 30–40 %. Siehe dort.
Wittig-ähnliche Reaktionen
Mit anderen Carbonyl- und Heterocarbonyl-Verbindungen
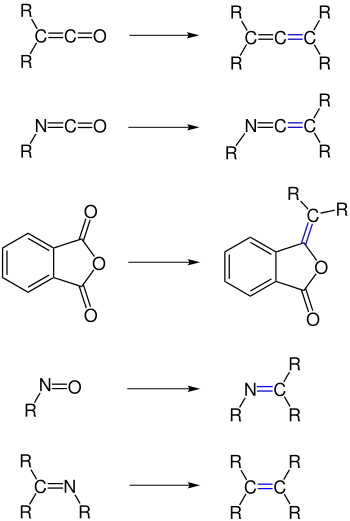
Phosphor-Ylide reagieren in ähnlicher Weise auch mit anderen Carbonylverbindungen als Aldehyden oder Ketonen, beispielsweise mit Ketenen,[22] mit Isocyanaten,[23] mit verschiedenen Anhydriden und Iminen.[24][25][26][27]
Arsen-Ylide
Das Homologe des Phosphors bildet wie dieses quartäre Arsonium-Salze, die sich ebenfalls mit Basen in α-Stellung deprotonieren lassen. Diese Arsen-Ylide verhalten sich bei der Carbonyl-Olefinierung wie Stickstoff-Ylide.[28]
Beispiele
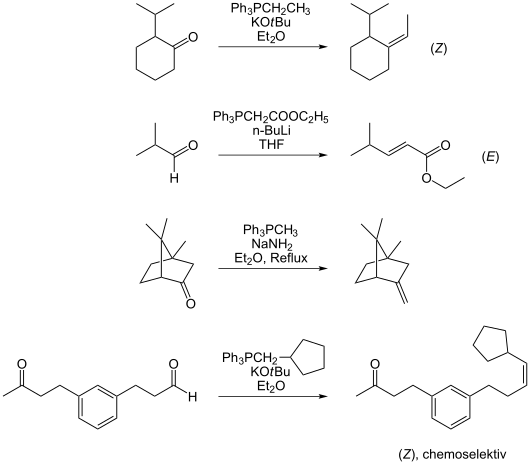
Die Vielseitigkeit der Wittig-Reaktion wird in folgender Abbildung an einigen Beispielen gezeigt:
Grenzen der Wittig-Reaktion
Ein Nachteil der Wittig-Reaktion ist ihre Beschränkung auf Aldehyde und Ketone (Ausnahmen siehe oben). Carbonsäurederivate sind quasi inert gegenüber Yliden. Die Wittig-Reaktion erfordert weiterhin basische Bedingungen, die Nebenreaktionen wie Eliminierungen oder Racemisierungen initiieren können. Alternative Olefinierungsreagenzien sind beispielsweise in Titanocen-Verbindungen (siehe Tebbe-Reaktion).
Alternativen der Wittig-Reaktion
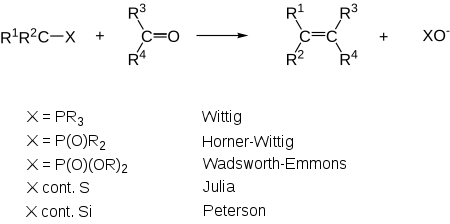
Horner-Wadsworth-Emmons-Reaktion, die Peterson-Olefinierung, die Julia-Olefinierung und die Tebbe-Reaktion. Ferner die Metathese.
Varianten
- Schlosser-Variante der Wittig-Reaktion
Literatur
- Thomas Laue, Andreas Plagens: Namens- und Schlagwortreaktionen der organischen Chemie. 5. Auflage. Vieweg+Teubner-Verlag, 2006, ISBN 3-8351-0091-2.
- Marcel Hoffmann: Entwicklung katalytischer Wittig-Reaktionen. Berlin, Mensch-&-Buch-Verlag, 2014, ISBN 978-3-86387-476-6.
Einzelnachweise
- G. Wittig, G. Geissler: Zur Reaktionsweise des Pentaphenyl-phosphors und einiger Derivate. In: Liebigs Ann. Chem. Band 580, Nr. 1, 1953, S. 44–57, doi:10.1002/jlac.19535800107.
- G. Wittig, U. Schöllkopf: Über Triphenyl-phosphin-methylene als olefinbildende Reagenzien. In: Chemische Berichte. Band 87, Nr. 9, 1954, S. 1318–1330, doi:10.1002/cber.19540870919.
- G. Wittig, H. Pommer: DBP 954247, 1956.
- G. Wittig, H. Pommer: In: Chem. Abstr. 53, 1959, S. 2279.
- Heinz G. Becker u. a. (Hrsg.): Organikum. Organisch-chemisches Grundpraktikum. Wiley-VCH, Weinheim 2004, ISBN 3-527-31148-3.
- L. Fitjer, U. Quabeck: In Chem. Commun. 15, 1985, S. 855–864.
- D. Spitzner, K. Oesterreich: Anionically Induced Domino Reactions − Synthesis of a Norpatchoulenol-Type Terpene. In: European Journal of Organic Chemistry. Band 2001, Nr. 10, 2001, S. 1883–1886, doi:10.1002/1099-0690(200105)2001:10<1883::AID-EJOC1883>3.0.CO;2-M.
- M. Schlosser, B. Schaub: In Chimia 36, 1982, S. 396–397.
- Jerry March: Advanced Organic Chemistry. McGraw-Hill, Kogakusha 1977, ISBN 0-07-040247-7.
- K. P. C. Vollhardt, N. E. Schore: Organische Chemie. 4. Auflage. Wiley-VCH, Weinheim 2005, ISBN 3-527-31380-X, S. 891.
- Robert B. Woodward, Roald Hoffmann: Die Erhaltung der Orbitalsymmetrie. Verlag Chemie, Weinheim 1970, ISBN 3-527-25323-8, S. 1–178.
- Kaim, W.: Einelektronenübertragung: Abschied von Elektronenpaar-Mechanismen? In: Nachr. Chem. Tech. Lab. 32, 1984, S. 436–439.
- Peter Sykes: Reaktionsmechanismen der Organischen Chemie. 7. Auflage. Verlag Chemie, 1979, ISBN 3-527-21047-4.
- M. Schlosser, K. F. Christmann In: Liebigs Ann. Chem. 708, 1967, S. 1.
- Thomas Laue, Andreas Plagens: Namens- und Schlagwortreaktionen der organischen Chemie. 5. Auflage. Vieweg+Teubner-Verlag, 2006, ISBN 3-8351-0091-2.
- B. E. Maryanoff, A. B. Reitz, M. S. Mutter, R. R. Whittle, R. A. Olofson: Stereochemistry and mechanism of the Wittig reaction. Diasteromeric reaction intermediates and analysis of the reaction course. In: J. Am. Chem. Soc. Band 108, Nr. 24, 1986, S. 7664–7678, doi:10.1021/ja00284a034.
- E. Vedejs u. a.: Topics in Stereochemistry. Band 21, 1994, ISBN 0-471-52120-5.
- A. Streitwieser, C. H. Heathcock: Organische Chemie. Verlag-Chemie, 1980, ISBN 3-527-25810-8.
- Nguyên Trong Anh: Die Woodward-Hoffmann-Regeln und ihre Anwendung. Verlag Chemie, 1970, ISBN 3-527-25430-7.
- H.-J. Bestmann, R. Armsen, H. Wagner, Chem. Ber. 102, 1969, S. 2259–2269.
- B. M. Trost, D. P. Curran: An enantiodirected cyclopentenone annulation. Synthesis of a useful building block for condensed cyclopentanoid natural products. In: J. Am. Chem. Soc. Band 102, Nr. 17, 1980, S. 5699–5700, doi:10.1021/ja00537a059.
- Asknes und Frøyen, Acta Chem. Scand., 22, 1968, S. 2347.
- Frøyen: Acta Chem. Scand., Ser. B. 28, 1974, S. 568.
- Chopard, Hudson, Searle: Tetrahedron Lett., 1965, S. 2357.
- Flitsch, Peters: Tetrahedron Lett., 1969, S. 1161.
- Gara, Massy-Westropp und Reynolds: Tetrahedron Lett., 1969, S. 4171.
- Bestmann, Seng: Tetrahedron. 21, 1965, S. 1373.
- A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 81.–90. Auflage. Walter de Gruyter, Berlin 1976, ISBN 3-11-005962-2.