SDS-PAGE
SDS-PAGE (Abkürzung für englisch sodium dodecyl sulfate polyacrylamide gel electrophoresis, Natriumdodecylsulfat-Polyacrylamidgelelektrophorese) ist eine Variante der Polyacrylamid-Gelelektrophorese, einer analytischen Methode der Biochemie zur Trennung von Stoffgemischen nach der Molekülmasse in einem elektrischen Feld.
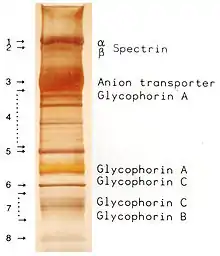
Dieses von Ulrich K. Laemmli entwickelte diskontinuierliche elektrophoretische System ermöglicht eine gute Trennung von Proteinen mit Molekülmassen zwischen 5 und 250 kDa.[1] Die Publikation, in der es beschrieben wurde, ist das am häufigsten zitierte Paper eines Einzelautors, und das am zweithäufigsten zitierte insgesamt.[2]
Eigenschaften

Die SDS-PAGE wird zur Analyse von Proteinen verwendet.[3] Als Trennmedium (auch als Matrix bezeichnet) bei dieser Art der Elektrophorese dient ein diskontinuierliches Gel auf Polyacrylamidbasis. Zusätzlich kommt SDS (Natriumdodecylsulfat) zum Einsatz. Dieses anionische Tensid (Detergens) überdeckt die Eigenladungen von Proteinen. Pro Gramm Protein binden konstant ungefähr 1,4 Gramm SDS,[4][5][6] entsprechend einem SDS-Molekül pro zwei Aminosäuren, sodass die Proteine eine konstante negative Ladungsverteilung aufweisen. Die Eigenladungen der Proteine sind unter der SDS-Beladung vernachlässigbar, die positiven Ladungen sind zudem im basischen pH-Wert-Bereich eines Trenngels nach Laemmli stark verringert. Die negativen Ladungen des SDS bewirken deren gegenseitige Abstoßung, was zusammen mit der Denaturierung durch Aufkochen zu einer Linearisierung der zuvor gefalteten Proteine führt. Dies erlaubt eine Auftrennung nach der Kettenlänge, proportional zur Molekülmasse, denn längere Proteine werden im Gel stärker zurückgehalten als kürzere.
SDS neigt in wässrigen Lösungen ab einer bestimmten Konzentration zur Bildung kugelförmiger Mizellen. SDS kommt in Lösungen ab der kritischen Mizellenbildungskonzentration von 7 bis 10 Millimolar gleichzeitig als einzelne Moleküle (Monomer) und als Mizellen vor, darunter kommt SDS nur einzeln vor. Bei der kritischen Mizellenbildungskonzentration besteht eine Mizelle aus etwa 62 SDS-Molekülen.[7] Allerdings binden nur SDS-Monomere über hydrophobe Wechselwirkungen an Proteine, während die auf der Außenseite anionischen SDS-Mizellen kein Protein aufnehmen.[5] In SDS-Konzentrationen über 0,1 Millimolar beginnt die Entfaltung von Proteinen,[5] oberhalb von 1 mM werden die meisten Proteine denaturiert. Wegen des starken Denaturierungseffekts von SDS und der folgenden Aufspaltung von Proteinkomplexen können in der Regel mit SDS keine Quartärstrukturen bestimmt werden. Ausnahmen bilden z. B. zuvor durch kovalente Quervernetzung stabilisierte Proteine und die SDS-resistenten Proteinkomplexe, die auch in Gegenwart von SDS stabil sind (Letztere jedoch nur bei Raumtemperatur). Die SDS-Resistenz basiert auf der Metastabilität: Um die resistenten Komplexe zu denaturieren, ist eine hohe Aktivierungsenergie erforderlich, die durch Erhitzen erreicht wird. Obwohl das native, vollständig gefaltete, SDS-resistente Protein unter den Bedingungen keine ausreichende Stabilität besitzt, stellt sich das chemische Gleichgewicht der Denaturierung bei Raumtemperatur nur langsam ein. Stabile Proteinkomplexe zeichnen sich neben der SDS-Resistenz auch noch durch Stabilität gegen Proteasen und eine erhöhte biologische Halbwertszeit aus.[8]
Alternativ kann teilweise auch eine Polyacrylamid-Gelelektrophorese mit den kationischen Tensiden CTAB in einer CTAB-PAGE[9][10][11] oder 16-BAC in einer BAC-PAGE verwendet werden.[12]
Physikalische Grundlagen
In einem elektrischen Feld wandern geladene Teilchen in Lösung zum Pol mit der entgegengesetzten Ladung. Durch die Beladung von Proteinen mit SDS sind sie mit mehreren negativen Ladungen behaftet, weshalb Proteine in der SDS-PAGE zum Plus-Pol wandern. Durch Verwendung eines ungeladenen polymeren Hydrogels (Polyacrylamid) kommt ein verstärkter Siebeffekt hinzu. Die Porengröße im Hydrogel wird durch die Konzentrationen an Monomer und Vernetzer bestimmt.
Verfahren
Das Verfahren der SDS-PAGE setzt sich aus der Gelherstellung, der Probenvorbereitung, der Elektrophorese, der Proteinfärbung oder einem Western Blot und der Analyse des erzeugten Bandenmusters zusammen.
Gelherstellung


Bei Verwendung unterschiedlicher Puffer im Gel (diskontinuierliche Gelelektrophorese) werden die Gele bis zu einem Tag vor der Elektrophorese hergestellt, damit die Diffusion nicht zu einer Vermischung der Puffer führt. Das Gel wird durch radikalische Polymerisation in einer Form erzeugt, die aus zwei abgedichteten Glasplatten mit Abstandshaltern zwischen den Glasplatten besteht. Die Abstandshalter besitzen eine Dicke von 0,75 mm oder 1,5 mm, welche die Ladekapazität des Gels bestimmt. Die Platten werden meistens für das Gießen der Gellösung in einen Ständer eingespannt, der die sonst offene Unterseite der Glasplatten mit den beiden Abstandshaltern vorübergehend abdichtet. Für die Gellösung werden Acrylamid als Gelbildner (meistens 4 % V/V im Sammelgel und 10–12 % im Trenngel), Methylenbisacrylamid als Quervernetzer, Sammel- oder Trenngelpuffer sowie Wasser und SDS gemischt. Durch Zugabe des Katalysators TEMED und des Radikalstarters Ammoniumpersulfat (APS) wird die Polymerisation begonnen. Die Lösung wird anschließend blasenfrei zwischen die Glasplatten gegossen. Je nach Menge an Katalysator und Radikalstarter sowie je nach Temperatur dauert die Polymerisation zwischen einer viertel Stunde und mehreren Stunden. Das untere Gel (Trenngel) wird zuerst gegossen und mit ein paar Tropfen von einem wenig wasserlöslichen Alkohol (meistens puffergesättigtes Butanol oder Isopropanol) überschichtet, wodurch der Meniskus blasenfrei wird und vor dem Radikalfänger Sauerstoff geschützt wird. Nach der Polymerisation des Trenngels wird der Alkohol abgekippt und die Reste mit Filterpapier entfernt. Nach Zugabe von APS und TEMED zur Sammelgellösung erfolgt das Gießen der Sammelgellösung auf das feste Trenngel und das blasenfreie Einsetzen eines geeigneten Probenkamms. Der Probenkamm wird nach der Polymerisation vorsichtig herausgezogen, wodurch Taschen für den Probenauftrag entstehen. Für eine spätere Verwendung der Proteine für eine Proteinsequenzierung werden die Gele oftmals am Vortag der Elektrophorese hergestellt, um Reaktionen von unpolymerisiertem Acrylamid mit Cysteinen in Proteinen zu mindern.
Durch Verwendung eines Gradientenmischers können Gradienten-Gele mit einem Gradienten des Acrylamids (meist von 4 bis 12 %) gegossen werden, die einen größeren Trennbereich der Molmassen aufweisen.[13] Kommerzielle Gelsysteme (so genannte pre-cast-Gele) verwenden meist die Puffersubstanz BisTris mit einem pH-Wert zwischen 6,4 und 7,2 sowohl im Sammel- als auch im Trenngel.[14][15] Diese Gele werden bereits fertig gegossen geliefert und sind umgehend einsatzbereit. Da sie nur einen Puffer verwenden (kontinuierliche Gelelektrophorese) und einen nahezu neutralen pH-Wert aufweisen, sind sie über mehrere Wochen lagerbar. Der neutralere pH-Wert verlangsamt die Hydrolyse und somit den Zerfall des Polyacrylamids.[16] Weiterhin kommt es zu weniger Acrylamid-modifizierten Cysteinen in den Proteinen.[14] Aufgrund des konstanten pH-Werts in Sammel- und Trenngel gibt es keinen Stapelungseffekt. Proteine in BisTris-Gelen können nicht mit Rutheniumkomplexen gefärbt werden.[17] Dieses Gelsystem besitzt einen vergleichsweise großen Auftrennungsbereich, der durch Verwendung von MES oder MOPS im Laufpuffer variiert werden kann.[14]
Probenvorbereitung

Bei der Probenvorbereitung wird Probenpuffer und somit SDS im Überschuss zu den Proteinen hinzugegeben und die Probe anschließend für fünf Minuten auf 95 °C erhitzt, um Sekundär- und Tertiärstrukturen durch das Unterbrechen von Wasserstoffbrücken und das Strecken der Moleküle aufzubrechen. Optional können Disulfidbrücken durch Reduktion gespalten werden. Dazu werden reduzierende Thiole wie β-Mercaptoethanol (β-ME, 5 % Volumenanteil), Dithiothreitol (DTT, 10 Millimolar) oder Dithioerythrit (DTE, 10 Millimolar) dem Probenpuffer zugesetzt. Jede Probe wird in ihre eigene Tasche im Gel pipettiert, das zuvor im Elektrophoreseapparat in Elektrophoresepuffer untergetaucht wurde.
Zusätzlich zu den Proben wird meistens ein Größenmarker auf das Gel geladen. Dieser besteht aus Proteinen von bekannter Größe und ermöglicht dadurch die Abschätzung der Größe der Proteine in den eigentlichen Proben mit einer Genauigkeit von ± 10 %,[18] die parallel in unterschiedlichen Spuren des Gels wandern. Der Größenmarker wird oftmals in die erste oder letzte Tasche eines Gels pipettiert.
Elektrophorese

Zur Auftrennung werden die denaturierten Proben auf ein Gel aus Polyacrylamid geladen, das in einen Elektrophoresepuffer mit geeigneten Elektrolyten eingelegt ist. Danach wird eine elektrische Spannung (meist um die 100 Volt, 10–20 V pro cm Gellänge) angelegt, die eine Migration der negativ geladenen Moleküle durch das Gel in Richtung der Anode (Plus-Pol) bewirkt. Das Gel wirkt dabei wie ein Sieb. Kleine Proteine wandern relativ leicht durch die Maschen des Gels, während große Proteine eher zurückgehalten werden und dadurch langsamer durch das Gel wandern. Die Elektrophorese dauert je nach verwendeter Spannung und Länge des Gels zwischen einer dreiviertel Stunde und mehreren Stunden.
Die am schnellsten wandernden Proteine (mit einer Molmasse von unter 5 KDa) bilden mit den ebenfalls durch das Gel wandernden anionischen Bestandteilen des Elektrophoresepuffers die Laufmittelfront. Der Bereich der Laufmittelfront wird durch den Zusatz des vergleichsweise kleinen, anionischen Farbstoffs Bromphenolblau zum Probenpuffer sichtbar gemacht. Aufgrund der relativ geringen Molekülgröße des Bromphenolblaus wandert es schneller als Proteine. Durch die optische Kontrolle anhand der wandernden farbigen Bande kann die Elektrophorese beendet werden, bevor der Farbstoff und somit auch die Proben das Gel vollständig durchwandert haben und es wieder verlassen.
Das am häufigsten eingesetzte Verfahren ist die diskontinuierliche SDS-PAGE. Bei dieser wandern die Proteine zuerst in ein Sammelgel mit neutralem pH, in dem sie konzentriert werden und anschließend in ein Trenngel mit basischem pH, in dem die eigentliche Auftrennung erfolgt. Sammel- und Trenngel unterscheiden sich durch unterschiedliche Porengröße (4–6%T bzw. 10-20%T), Ionenstärke und pH-Werte (pH6,8 bzw. pH8,8). Als Elektrolyt wird häufig ein SDS-haltiges TRIS-Glycin-Chlorid-Puffersystem eingesetzt. Glycin bildet bei neutralem pH-Wert überwiegend die zwitterionische Form aus, bei hohen pH-Werten verlieren die Glycinationen positive Ladungen und werden vorwiegend anionisch. Im Sammelgel wandern die kleineren, negativ geladenen Chloridionen vor den Proteinen (engl. leading ions ‚führende Ionen‘) und die etwas größeren, negativ und teilweise positiv geladenen Glycinat-Ionen nach (engl. trailing ions ‚folgende Ionen‘), während im vergleichsweise basischen Trenngel beide Ionen vor den Proteinen wandern. Der pH-Gradient zwischen Sammel- und Trenngelpuffer führt zu einem Stapelungseffekt an der Grenze des Sammelgels zum Trenngel, da das Glycinat beim ansteigenden pH-Wert teilweise seine bremsende positive Ladung verliert und dann als zuvor folgendes Ion die Proteine beim Wandern überholt und zum führenden Ion wird, wodurch die nach einer Färbung sichtbaren Banden der verschiedenen Proteine schmaler und schärfer werden (Stapelungseffekt). Zur Trennung von kleineren Proteinen und Peptiden eignet sich aufgrund der höheren Spreizung der Proteine im Bereich von 0,5 bis 50 KDa das TRIS-Tricin-Puffersystem von Schägger und von Jagow.[19]
Färbung
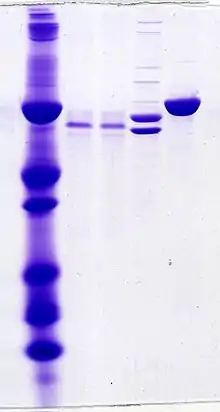
Am Ende der elektrophoretischen Trennung sind alle Proteine nach Größe sortiert und können anschließend durch weitere Verfahren (Proteinfärbungen wie z. B. die Coomassiefärbung (einfache Durchführung, am häufigsten verwendet),[20][21] die Silberfärbung (höchste Sensitivität),[22][23][24][25][26][27] die Stains-all-Färbung, die Amidoschwarz 10 B-Färbung,[21] die Fast-Green-FCF-Färbung,[21] die fluoreszenten Färbungen wie die Epicocconon-Färbung[28] und die SYPRO-Orange-Färbung[29] sowie immunologische Nachweise wie z. B. beim Western Blot) sichtbar gemacht werden.[30][31] Die fluoreszenten Färbungen besitzen eine vergleichsweise höhere Linearität zwischen Proteinmenge und Farbintensität von etwa drei Zehnerpotenzen oberhalb der Nachweisgrenze, d. h. man kann ungefähr aus der Farbintensität die Menge an Protein abschätzen. Bei einer Verwendung des fluoreszenten Proteinfarbstoffs Trichlorethanol entfällt eine nachträgliche Proteinfärbung, wenn es zur Gellösung hinzugefügt wurde und das Gel nach der Elektrophorese mit UV-Licht bestrahlt wurde.[32][33]
Analyse
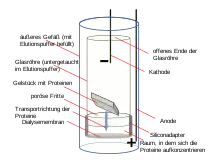
Durch eine Proteinfärbung erhält das Gel mit den getrennten Proteinen ein dokumentierbares Bandenmuster der verschiedenen Proteine. Glykoproteine adsorbieren SDS an den Glykosylierungen ungleichmäßiger, was in breiteren und unschärferen Banden resultiert.[34] Membranproteine sind wegen ihrer Transmembrandomäne häufig aus den hydrophoberen Aminosäuren aufgebaut, besitzen eine geringere Löslichkeit in wässrigen Lösungen, neigen zur Bindung von Lipiden und neigen aufgrund hydrophober Effekte zum Ausfallen in wässrigen Lösungen, wenn nicht ausreichend Detergens vorhanden ist. Dieses Ausfallen äußert sich bei Membranproteinen in der SDS-PAGE in einer „Schweifbildung“ oberhalb der Bande des Transmembranproteins, dagegen kann mehr SDS (durch Einsatz von mehr oder konzentrierterem Probenpuffer) verwendet und die Proteinmenge bei der Gelauftragung gemindert werden. Eine Überladung des Gels mit einem löslichen Protein äußert sich in einer halbkreisförmigen Bande dieses Proteins (z. B. in der Markerspur der Abbildung bei 66 KDa), wodurch andere Proteine mit ähnlichen Molmassen überdeckt werden können. Ein geringer Kontrast (wie in der Markerspur der Abbildung) zwischen den Banden innerhalb einer Spur deutet entweder auf das Vorhandensein vieler Proteine mit dazwischenliegenden Molmassen hin (geringe Reinheit) oder, wenn bei gereinigten Proteinen ein mangelnder Kontrast nur unterhalb einer Bande vorkommt, auf eine proteolytische Degradation des Proteins hin, die sich zuerst in Abbaubanden und nach weiterer Degradation auch in einer homogenen Färbung („Schmier“) unterhalb einer Bande äußert.[35] Die Dokumentation des Bandenmusters erfolgt meistens durch Fotografieren oder Scannen. Zur anschließenden Rückgewinnung der Moleküle in einzelnen Banden kann eine Gelextraktion durchgeführt werden. Zum Herauslösen der Proteine aus dem Gelstück wir auch häufig eine Elektroelution durchgeführt, bei der elektrophoretisch mithilfe eines elektrischen Feldes die SDS-beladenen Proteine aus dem Gel in eine Lösung transportiert werden. Hierzu werden spezielle Elektroelutionskammern benutzt.
Archivierung

Nach der Proteinfärbung und Dokumentation des Bandenmusters kann das Polyacrylamidgel für eine Archivierung getrocknet werden. Proteine können daraus zu einem späteren Zeitpunkt extrahiert werden. Dabei wird das Gel entweder in einen Trockenrahmen (mit oder ohne Wärmezufuhr) oder in einen Vakuumgeltrockner eingelegt. Der Trockenrahmen besteht aus zwei Teilen, von denen einer als Unterlage für eine nasse Zellophanfolie dient, auf die das Gel und eine einprozentige Glycerollösung gegeben wird. Darauf wird eine zweite nasse Zellophanfolie blasenfrei aufgelegt, der zweite Rahmenteil aufgesetzt und der Rahmen mit ein paar Klammern abgedichtet. Die Entfernung der Luftblasen vermeidet eine Fragmentierung des Gels beim Trocknen. Das enthaltene Wasser verdampft durch die Zellophanfolie. Ein Vakuumgeltrockner erzeugt dagegen einen Unterdruck und erwärmt das Gel auf etwa 50 °C. Ein Geltrockenofen verwendet ein Warmluftgebläse.
Molmassenbestimmung

Bei einer exakteren Bestimmung der Molmasse werden die relativen Laufweiten der einzelnen Proteinbanden im Trenngel gemessen.[36][37] Die Messungen erfolgen meist in Triplikaten zur Erhöhung der Genauigkeit. Die relative Laufweite (auch Rf-Wert, Rm-Wert) ist der Quotient aus der Laufweite der Bande des betrachteten Proteins und der Laufweite der Proteinfront. Die Laufweiten der Banden und der Proteinfront werden jeweils ab dem Beginn des Trenngels gemessen. Die Laufweite der Proteinfront entspricht ungefähr der Laufweite des im Probenpuffer enthaltenen Bromphenolblaus. Die relativen Laufweiten der Proteine des Größenmarkers werden halblogarithmisch gegen ihre bekannten Molmassen aufgetragen. Über Vergleich mit dem linearen Bereich des erzeugten Graphen oder rechnerisch durch eine Regressionsanalyse kann die Molmasse eines unbekannten Proteins anhand seiner relativen Laufweite ermittelt werden. Banden von Proteinen mit Glykosylierungen können unschärfer sein. Proteine mit vielen basischen Aminosäuren (z. B. Histone)[38] können zu einer Überschätzung der Molmasse führen oder gar nicht erst ins Gel einwandern, da sie bei der Elektrophorese wegen der positiven Ladungen langsamer oder in die entgegengesetzte Richtung wandern. Entsprechend können viele saure Aminosäuren zu einer beschleunigten Wanderung eines Proteins und einer Unterschätzung der Molmasse führen.[39]
Anwendungen
Die SDS-PAGE in Kombination mit einer Proteinfärbung wird in der Biochemie zur schnellen und genauen Trennung und anschließenden Analyse von Proteinen verwendet. Sie hat vergleichsweise geringe Geräte- und Reagenzkosten und ist eine vergleichsweise einfache Methode. Aufgrund der geringen Skalierbarkeit wird sie hauptsächlich für analytische Zwecke und weniger für präparative Zwecke verwendet, insbesondere wenn größere Mengen eines Proteins isoliert werden sollen.
Zusätzlich wird die SDS-PAGE in Kombination mit dem Western Blot zur Bestimmung des Vorhandenseins eines spezifischen Proteins in einer Mischung von Proteinen verwendet – oder für die Analyse von posttranslationalen Modifikationen. Posttranslationale Modifikationen von Proteinen können zu einer unterschiedlichen relativen Mobilität (d. h. eine Bandenverschiebung) oder zu einer Änderung der Bindung eines Nachweisantikörpers führen, der im Western-Blot verwendet wird (d. h. eine Bande verschwindet oder erscheint).
In der Massenspektrometrie von Proteinen, ist die SDS-PAGE eine weit verbreitete Methode für die Probenvorbereitung vor der Spektrometrie, meistens mit einem in-Gel-Verdau. In Bezug auf die Bestimmung der molekularen Masse eines Proteins ist die SDS-PAGE etwas genauer als eine analytische Ultrazentrifugation, aber weniger genau als eine Massenspektrometrie oder – ohne Berücksichtigung posttranslationaler Modifikationen – eine Berechnung der Proteinmolekülmasse aus der DNA-Sequenz.
In der medizinischen Diagnostik wird die SDS-PAGE unter anderem im Rahmen eines HIV-Tests sowie zur Untersuchung einer Proteinurie verwendet. Beim HIV-Test werden HIV-Proteine per SDS-PAGE getrennt und anschließend per Western Blot mit HIV-spezifischen Antikörpern des Patienten nachgewiesen, sofern sie in seinem Blutserum vorhanden sind. Bei einer SDS-PAGE zur Proteinurie werden die Mengen verschiedener Serumproteine im Urin bewertet, z. B. Albumin, Alpha-2-Makroglobulin und IgG.
Varianten
Die SDS-PAGE ist die am meisten verwendete Methode zur gelelektrophoretischen Trennung von Proteinen. Die 2D-Gelelektrophorese kombiniert nacheinander eine isoelektrische Fokussierung oder eine BAC-PAGE mit einer SDS-PAGE. Die Nativ-PAGE wird eingesetzt, wenn die native Proteinfaltung erhalten bleiben soll. Zur Trennung von Membranproteinen kann alternativ zur SDS-PAGE eine BAC-PAGE oder eine CTAB-PAGE verwendet werden. Zur elektrophoretischen Trennung von größeren Proteinkomplexen kann eine Agarose-Gelelektrophorese eingesetzt werden, z. B. die SDD-AGE. Enzyme können teilweise über ihre Enzymaktivität per Zymographie nachgewiesen werden.
Alternativen
Während die SDS-PAGE eine der präziseren und kostengünstigeren Methoden zur Proteintrennung und -analyse ist, denaturiert sie Proteine. Wo nicht-denaturierende Bedingungen erforderlich sind, werden Proteine zum Beispiel durch eine native PAGE oder verschiedene chromatographische Verfahren mit anschließender photometrischer Mengenbestimmung untersucht, wie die Affinitätschromatographie (oder sogar eine Tandem Affinity Purification), Größenausschlusschromatographie oder die Ionenaustauschchromatographie.[40] Proteine können auch nach Größe in einer Tangential-Flow-Filtration[41] oder einer Ultrafiltration separiert werden.[42] Einzelne Proteine können aus einer Mischung durch Affinitätschromatographie oder durch einen Pull-Down-Assay isoliert werden. Einige historisch frühe und kosteneffektive, aber unsaubere Trennungsmethoden, die üblicherweise auf einer Reihe von Extraktionen und Fällungen beruhen und kosmotrope Moleküle verwenden, sind zum Beispiel die Ammoniumsulfat-Fällung und die Polyethylenglykol-Fällung. Aufgrund der geringen Kosten werden sie heute noch zur Reinigung größerer Mengen von Proteinen verwendet.
Geschichte
Für die Entdeckung des Prinzips der Elektrophorese als Wanderung geladener und gelöster Atome oder Moleküle in einem elektrischen Feld wurde Arne Tiselius 1948 der Nobelpreis für Chemie verliehen.[43] Die Verwendung einer festen Matrix (anfänglich Papierscheiben) in einer Zonenelektrophorese verbesserte die Auftrennung. Die diskontinuierliche Elektrophorese aus dem Jahr 1964 von L. Ornstein und B. J. Davis ermöglichte die Verbesserung der Auftrennung durch den Stapelungseffekt.[44] Die Verwendung quervernetzter Polyacrylamid-Hydrogele bot im Gegensatz zu den zuvor verwendeten Papierscheiben oder Stärkegelen eine höhere Stabilität des Gels und keine mikrobielle Zersetzung. Die denaturierende Wirkung des SDS in kontinuierlichen Polyacrylamidgelen und die daraus folgende Verbesserung der Auftrennung wurde erstmals 1965 durch David F. Summers in der Arbeitsgruppe von James E. Darnell für die Auftrennung der Proteine des Poliovirus beschrieben.[45] Die heutige Variante der SDS-PAGE wurde 1970 von Ulrich K. Laemmli beschrieben und erstmals zur Charakterisierung der Proteine im Kopf des Bakteriophagen T4 verwendet.[1]
Literatur
- Hubert Rehm, Thomas Letzel: Der Experimentator: Proteinbiochemie / Proteomics. 6. Auflage, Spektrum Akademischer Verlag, Heidelberg 2009, ISBN 978-3-8274-2312-2.
Weblinks
- OpenWetWare: Protokoll für BisTris SDS-PAGE
Einzelnachweise
- U. K. Laemmli: Cleavage of structural proteins during the assembly of the head of bacteriophage T4. In: Nature. Bd. 227, 1970, S. 680–685, doi:10.1038/227680a0, PMID 5432063.
- Interview mit Ulrich Laemmli. In: NZZ Folio. Nr. 11, 2005. Abgerufen am 4. März 2012.
- B. J. Smith: SDS Polyacrylamide Gel Electrophoresis of Proteins. In: Methods in molecular biology (Clifton, N.J.). Band 1, 1984, S. 41–55, doi:10.1385/0-89603-062-8:41, PMID 20512673.
- R. Pitt-Rivers, F. S. Impiombato: The binding of sodium dodecyl sulphate to various proteins. In: The Biochemical journal. Band 109, Nummer 5, Oktober 1968, S. 825–830, PMID 4177067, PMC 1187034 (freier Volltext).
- J. A. Reynolds, Charles Tanford: Binding of dodecyl sulfate to proteins at high binding ratios. Possible implications for the state of proteins in biological membranes. In: Proceedings of the National Academy of Sciences. Band 66, Nummer 3, Juli 1970, S. 1002–1007, PMID 5269225, PMC 283150 (freier Volltext).
- Georgios Staikos, Anastasios Dondos: Study of the sodium dodecyl sulphate–protein complexes: evidence of their wormlike conformation by treating them as random coil polymers. In: Colloid and Polymer Science. 287, 2009, S. 1001, doi:10.1007/s00396-009-2059-3.
- Nicholas J. Turro, Ahmad Yekta: Luminescent probes for detergent solutions. A simple procedure for determination of the mean aggregation number of micelles. In: Journal of the American Chemical Society. 100, 1978, S. 5951, doi:10.1021/ja00486a062.
- Marta Manning, Wilfredo Colón: Structural basis of protein kinetic stability: resistance to sodium dodecyl sulfate suggests a central role for rigidity and a bias toward beta-sheet structure. In: Biochemistry. Bd. 43, 2004, S. 11248–11254, PMID 15366934.
- Engelbert Buxbaum: Cationic electrophoresis and electrotransfer of membrane glycoproteins. In: Analytical Biochemistry. Bd. 314, Nr. 1, 2003, S. 70–76, PMID 12633604, doi:10.1016/S0003-2697(02)00639-5.
- Dianne T. Akin, Raymond Shapira, Joseph M. Kinkade Jr.: The determination of molecular weights of biologically active proteins by cetyltrimethylammonium bromide-polyacrylamide gel electrophoresis. In: Analytical Biochemistry. Bd. 145, Nr. 1, 1985, S. 170–176, PMID 4003759, doi:10.1016/0003-2697(85)90343-4.
- R. J. Simpson: CTAB-PAGE. In: Cold Spring Harbor protocols. Band 2010, Nummer 4, April 2010, S. pdb.prot5412, doi:10.1101/pdb.prot5412, PMID 20360366.
- Joachim Hartinger, Katinka Stenius, Dagmar Högemann, Reinhard Jahn: 16-BAC/SDS-PAGE: a two-dimensional gel electrophoresis system suitable for the separation of integral membrane Proteins. In: Analytical Biochemistry. Bd. 240, Nr. 1, 1996, S. 126–133, PMID 8811889, doi:10.1006/abio.1996.0339.
- J. Margolis, K. G. Kenrick: 2-dimensional resolution of plasma proteins by combination of polyacrylamide disc and gradient gel electrophoresis. In: Nature. Band 221, Nummer 5185, März 1969, S. 1056–1057, PMID 5774398.
- J. P. Hachmann, J. W. Amshey: Models of protein modification in Tris-glycine and neutral pH Bis-Tris gels during electrophoresis: effect of gel pH. In: Analytical biochemistry. Band 342, Nummer 2, Juli 2005, S. 237–245, doi:10.1016/j.ab.2005.04.015, PMID 15935323.
- J. Wiltfang, N. Arold, V. Neuhoff: A new multiphasic buffer system for sodium dodecyl sulfate-polyacrylamide gel electrophoresis of proteins and peptides with molecular masses 100,000-1000, and their detection with picomolar sensitivity. In: Electrophoresis. Band 12, Nummer 5, Mai 1991, S. 352–366, doi:10.1002/elps.1150120507, PMID 1718736.
- A. Penna, M. Cahalan: Western Blotting using the Invitrogen NuPage Novex Bis Tris minigels. In: Journal of visualized experiments : JoVE. Nummer 7, 2007, S. 264, doi:10.3791/264, PMID 18989435, PMC 2565856 (freier Volltext).
- J. Moebius, K. Denker, A. Sickmann: Ruthenium (II) tris-bathophenanthroline disulfonate is well suitable for Tris-Glycine PAGE but not for Bis-Tris gels. In: Proteomics. Band 7, Nummer 4, Februar 2007, S. 524–527, doi:10.1002/pmic.200600642, PMID 17309097.
- Ian M. Rosenberg: Protein Analysis and Purification: Benchtop Techniques. Springer Science & Business Media, 22 December 2006, ISBN 978-0-8176-4412-3, S. 103.
- Hermann Schägger, Gebhard von Jagow: Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. In: Analytical Biochemistry. Bd. 166, 1987, S. 368–379, doi:10.1016/0003-2697(87)90587-2, PMID 2449095.
- S. Fazekas de St. Groth, R. G. Webster, A. Datyner: Two new staining procedures for quantitative estimation of proteins on electrophoretic strips. In: Biochimica et Biophysica Acta. 71, 1963, S. 377–391. doi:10.1016/0006-3002(63)91092-8. PMID 18421828.
- Wilson CM: Studies and critique of Amido Black 10B, Coomassie Blue R, and Fast Green FCF as stains for proteins after polyacrylamide gel electrophoresis.. In: Anal Biochem. 96, Nr. 2, 1979, S. 263-78. PMID 89822.
- C. R. Merril, R. C. Switzer, M. L. Van Keuren: Trace polypeptides in cellular extracts and human body fluids detected by two-dimensional electrophoresis and a highly sensitive silver stain. In: Proc Natl Acad Sci U S A. 76(9), 1979, S. 4335–4339. PMID 92027.
- R. C. Switzer, C. R. Merril, S. Shifrin: A highly sensitive silver stain for detecting proteins and peptides in polyacrylamide gels. In: Anal Biochem. Band 98, Nr. 1, September 1979, S. 231–237, doi:10.1016/0003-2697(79)90732-2, PMID 94518.
- T. Rabilloud u. a.: Improvement and simplification of low-background silver staining of proteins by using sodium dithionite. In: Electrophoresis. 9(6), 1998, S. 288–291. PMID 2466660.
- T. Rabilloud: A comparison between low background silver diammine and silver nitrate protein stains. In: Electrophoresis. 13, 1992, S. 429–439. PMID 1425556.
- C. Lelong, M. Chevallet, S. Luche, T. Rabilloud: Silver staining of proteins in 2DE gels. In: Methods Mol Biol. 519, 2009, S. 339–350. PMID 19381593.
- H. Blum, H. Beier, H. J. Gross: Improved silver staining of plant protein, RNA & DNA in PAA gels. In: Electrophoresis. 8, 1987, S. 93–99.
- Christian P. Moritz, Sabrina X. Marz, Ralph Reiss, Thomas Schulenborg, Eckhard Friauf: Epicocconone staining: a powerful loading control for Western blots. In: Proteomics. PMID 24339236.
- Aleksandr Petrovich Demchenko: Advanced Fluorescence Reporters in Chemistry and Biology III: Applications in Sensing and Imaging Band 3 von Advanced Fluorescence Reporters in Chemistry and Biology. Springer 2011, ISBN 978-3-642-18035-4, S. 179ff.
- S. Gallagher, D. Chakavarti: Staining proteins in gels. In: Journal of visualized experiments : JoVE. Nummer 17, 2008, ISSN 1940-087X, 760, doi:10.3791/760, PMID 19066521. PMC 3253607 (freier Volltext).
- C. M. Wilson: Staining of proteins on gels: comparisons of dyes and procedures. In: Methods in enzymology. Band 91, 1983, ISSN 0076-6879, S. 236–247, PMID 6190068.
- Carol L. Ladner, Jing Yang, Raymond J. Turner, Robert A. Edwards: Visible fluorescent detection of proteins in polyacrylamide gels without staining. In: Analytical Biochemistry Bd. 326, 2004, S. 13–20, PMID 14769330.
- Jennifer E. Gilda, Aldrin V. Gomes: Stain-Free total protein staining is a superior loading control to β-actin for Western blots. In: Analytical Biochemistry Bd. 440, 2013, S. 186–188, PMID 23747530.
- Cryo-EM Part A: Sample Preparation and Data Collection. Academic Press, 30 September 2010, ISBN 978-0-08-095695-4, S. 28–.
- Richard R Burgess, Murray P. Deutscher: Guide to Protein Purification. Academic Press, 3 November 2009, ISBN 978-0-08-092317-8, S. 184.
- Philip L.R. Bonner, Alan J. Hargreaves: Basic Bioscience Laboratory Techniques: A Pocket Guide. John Wiley & Sons, 24 August 2011, ISBN 978-1-119-95644-0, S. 140.
- Martin Holtzhauer: Basic Methods for the Biochemical Lab. Springer Science & Business Media, 13 September 2006, ISBN 978-3-540-32786-8, S. 243.
- W.J. van Venrooij, Ravinder N. Maini: Manual of Biological Markers of Disease. Springer Science & Business Media, 6 December 2012, ISBN 978-94-011-1670-1, S. 50.
- Y. Guan, Q. Zhu, D. Huang, S. Zhao, L. Jan Lo, J. Peng: An equation to estimate the difference between theoretically predicted and SDS PAGE-displayed molecular weights for an acidic peptide. In: Scientific reports. Band 5, 2015, S. 13370, doi:10.1038/srep13370, PMID 26311515, PMC 4550835 (freier Volltext).
- Jan-Christer Janson: Protein Purification: Principles, High Resolution Methods, and Applications. John Wiley & Sons, 3 January 2012, ISBN 978-1-118-00219-3.
- Mohamed A. Desai: Downstream Processing of Proteins: Methods and Protocols. Springer Science & Business Media, 2000, ISBN 978-1-59259-027-8, S. 35.
- Ghosh Raja: Protein Bioseparation Using Ultrafiltration: Theory, Applications And New Developments. World Scientific, 11 June 2003, ISBN 978-1-78326-126-0, S. 142.
- T. Pederson: Turning a PAGE: the overnight sensation of SDS-polyacrylamide gel electrophoresis. In: FASEB journal : official publication of the Federation of American Societies for Experimental Biology. Band 22, Nummer 4, April 2008, S. 949–953, doi:10.1096/fj.08-0402ufm, PMID 18378803.
- L. Ornstein, B. J. Davis: Disc Electrophoresis –1. Background and Theory. In: Ann NY Acad Sci. Band 121, 1964, S. 321–349.
- David F. Summers, Jacob V. Maizel, James E. Darnell: Evidence for virus-specific noncapsid proteins in poliovirus-infected HeLa cells. In: Proceedings of the National Academy of Sciences. Band 54, Nummer 2, August 1965, S. 505–513, PMID 4285933, PMC 219696 (freier Volltext).