Proteinfaltung
Die Proteinfaltung ist der Prozess, durch den Proteine ihre dreidimensionale Struktur erhalten. Sie findet während und nach der Synthese der Peptidkette statt und ist Voraussetzung für die fehlerfreie Funktion des Proteins. Bewirkt wird die Faltung durch kleinste Bewegungen der Lösungsmittelmoleküle (Wassermoleküle) und durch elektrische Anziehungskräfte innerhalb des Proteinmoleküls. Einige Proteine können nur mithilfe von bestimmten Enzymen oder Chaperon-Proteinen die richtige Faltung erreichen.
| Übergeordnet |
| Metabolismus der Proteine |
| Untergeordnet |
| de novo-Proteinfaltung Proteinfaltung mit Chaperonen |
| Gene Ontology |
|---|
| QuickGO |
Proteinsynthese
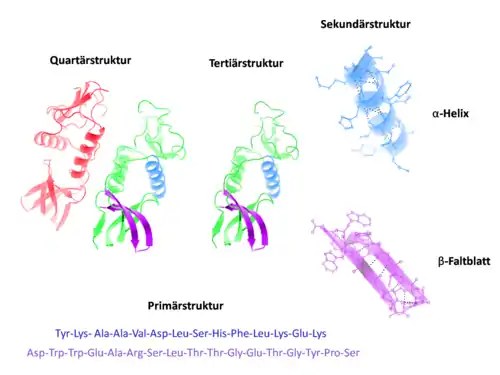
Proteine werden an den Ribosomen als lineare Polypeptidketten aus Aminosäuren synthetisiert. Die Abfolge der einzelnen Aminosäuren bildet die Primärstruktur des Proteins. Während oder nach der Synthese faltet sich die Polypeptidkette in eine definierte räumliche Struktur (Tertiärstruktur), die aus kleineren Strukturelementen (Sekundärstruktur) aufgebaut ist. Einige Proteine bestehen aus mehr als einer Polypeptidkette. Bildet sich ein solcher Oligomer aus mehreren Polypeptidketten in Tertiärstruktur, so spricht man von einer Quartärstruktur. Die Primärstruktur besteht aus kovalent über Peptidbindungen verbundenen Aminosäuren. Die Proteinfaltung ergibt sich aus den weiterhin wirkenden Bindungen und Kräften (ionische, polare und Van-der-Waals-Wechselwirkungen, Wasserstoffbrückenbindungen, hydrophobe Effekte) zwischen verschiedenen Atomen, in Einzelfällen wird die Sekundär-, Tertiär- und Quartärstruktur nach der Translation durch kovalente Bindungen wie Disulfidbrücken und Isopeptidbindungen stabilisiert.
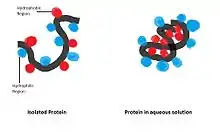
Die Aminosäurekette nimmt während der Faltung in Sekundenbruchteilen die native Konformation, also die biologisch funktionsfähige Konformation an. Dieser Prozess wird als Levinthal-Paradox bezeichnet. Das fertig gefaltete Protein hat in der Regel die niedrigste mögliche Gibbs-Energie (Anfinsen-Dogma). Der genaue Ablauf der Proteinfaltung ist noch nicht geklärt und stellt einen aktuellen Forschungsgegenstand der Biochemie dar. Bei einigen Proteinen verläuft die Faltung über einen Zwischenzustand, der molten globule genannt wird. Es kommt zu einer Zusammenlagerung der hydrophoben Aminosäurereste per hydrophobem Kollaps, wobei innerhalb von einigen Millisekunden alle Sekundärstrukturelemente ausgebildet werden. Erst im Anschluss kommt es zur Ausbildung der Tertiärstruktur, die einige Sekunden in Anspruch nehmen kann.
Faltungshelfer
Häufig können sich neu synthetisierte Proteine nicht selbst korrekt falten, daher werden Faltungshelfer benötigt. Chaperone verhindern durch Anlagerung die vorzeitige Proteinfaltung und somit die Aggregation während der Proteinsynthese. Chaperonine können Peptidsequenzen wie in einem Fass einschließen und durch ATP bei der Faltung helfen.
Protein-Disulfid-Isomerasen können falsch entstandene Disulfidbrücken korrigieren. Peptidyl-Prolyl-cis/trans-Isomerasen helfen dabei, Prolin von der cis-Konfiguration in eine trans-Konfiguration zu überführen. Andere Aminosäuren liegen zum großen Teil immer in trans-Konfiguration vor.[1]
Struktur und Funktion
Die spezifische Funktion eines Proteins ist nur durch seine definierte Struktur möglich. Fehlgefaltete Proteine werden normalerweise im Rahmen der Proteinqualitätskontrolle erkannt und im Proteasom abgebaut. Schlägt dieser Abbau fehl, kommt es zu Proteinansammlungen, die je nach Protein verschiedene Erkrankungen auslösen können, bei denen Mutationen die korrekte Faltung verhindern. Diese werden als Proteinfehlfaltungserkrankungen bezeichnet und können folgende Ursachen haben:
- Das Protein funktioniert nicht mehr. Beispiele: Formen von Krebs, die auf Mutationen im Protein p53 zurückgehen.
- Das Protein aggregiert. Beispiele: Sichelzellenanämie, bei der Hämoglobin aggregiert; Alzheimer-Krankheit; Parkinson-Krankheit; Chorea Huntington.
- Das Protein wirkt toxisch. Beispiel: BSE, verursacht durch die Prionen.
Denaturierung
Bei der Faltung nimmt das Protein den nativen (strukturierten oder biologisch funktionsfähigen) Zustand an. Den umgekehrten Prozess nennt man Denaturierung. Faltung und Denaturierung von Proteinen ähneln Phasenübergängen erster Ordnung, das heißt extensive Größen wie Volumen und Wärmeenergie ändern sich sprunghaft. Die Denaturierung von Proteinen wird beispielsweise durch Hitze, extreme pH-Bedingungen oder extreme Salzkonzentrationen ausgelöst.
1972 erhielt Christian B. Anfinsen den Chemie-Nobelpreis für die Beobachtung, dass sich kleine Proteine nach einer Denaturierung wieder in die native Form falten können, sobald sie Umgebungsbedingungen ausgesetzt werden, in denen diese stabil ist. Anfinsen schlussfolgerte aus dieser Beobachtung, dass die native Struktur eines jeden Proteins durch seine Aminosäuresequenz festgelegt ist.[2] Eine Ausnahme bilden seltene metamorphe Proteine wie Lymphotactin, die zwei grundsätzlich verschiedene Sekundärstrukturen haben.[3]
Forschung
Die erste umfassende Theorie zur Proteinfaltung wurde in den 1920er Jahren vom chinesischen Wissenschaftler Hsien Wu entwickelt. Im europäisch-amerikanischen Raum wurden die ersten wesentlichen Arbeiten von Christian B. Anfinsen (Nobelpreis für Chemie 1972) und Charles Tanford (Tanfordübergang) in den 1950er Jahren durchgeführt.
Zurzeit läuft das Projekt „Folding@home“ der Stanford-Universität zur Simulation dieser Faltungen, bei dem Internetnutzer mithelfen können, indem sie Rechenleistung zur Verfügung stellen. Auch die Projekte „POEM@home“ der Universität Karlsruhe und „Rosetta@home“ der University of Washington verfolgen dieses Ziel. Alle drei Projekte verwenden unterschiedliche Ansätze zur Simulation der Proteinfaltung. Das Verteilte System Folding@home wurde im März 2020 das erste Computing-System das ein exaFLOPS erreicht.[4][5][6][7] Einen weiteren Ansatz verfolgt das Computerspiel Foldit, bei dem die Spieler versuchen, ein Protein möglichst geschickt zu falten und es so auf ein niedriges Energieniveau zu bringen.
Des Weiteren findet alle zwei Jahre das Gemeinschaftsexperiment CASP statt, welches Forschergruppen die Möglichkeit bietet, die Qualität ihrer Methoden zur Vorhersage von Proteinstrukturen ausgehend von der Primärstruktur zu testen und sich einen Überblick über den aktuellen Stand auf diesem Forschungsgebiet zu verschaffen. Stand 2020 erlaubt Deep Learning die bisher genauesten Proteinstrukturvorhersagen[8], siehe auch AlphaFold.
Bislang gibt es zwei von der Wissenschaft anerkannte Modelle zur Proteinfaltung:
- Diffusions-Kollisions-Modell, bei dem zuerst ein Proteinkern (Nucleus) geformt wird und anschließend die Sekundärstruktur;
- Nukleations-Kondensations-Modell, bei dem die Sekundär- und Tertiärstruktur simultan gebildet werden.
Literatur
- F. Sterpone, G. Stirnemann, D. Laage: Magnitude and molecular origin of water slowdown next to a protein. In: Journal of the American Chemical Society. Band 134, Nummer 9, März 2012, S. 4116–4119. doi:10.1021/ja3007897. PMID 22335572.
Weblinks
- Gerd Ulrich Nienhaus: Physik der Proteine. In: Physik Journal. Band 3, Nr. 4, 2004, S. 37–43 (pro-physik.de [PDF; abgerufen am 1. September 2020]). Strukturdynamik gefalteter Proteine
- AlphaFold. Strukturvorhersagen und Datenbank von Proteinstrukturen. In: European Bioinformatics Institute. Abgerufen am 24. Juli 2021 (englisch).
Einzelnachweise
- Proteinfaltung - Chemgapedia. Abgerufen am 4. Februar 2020.
- Horton, Robert et al.: Biochemie, 4. Auflage, Pearson Studium, München (2008), S. 146., ISBN 978-3-8273-7312-0.
- R. L. Tuinstra, F. C. Peterson, S. Kutlesa, E. S. Elgin, M. A. Kron, B. F. Volkman: Interconversion between two unrelated protein folds in the lymphotactin native state. In: Proceedings of the National Academy of Sciences. Band 105, Nummer 13, April 2008, S. 5057–5062, ISSN 1091-6490. doi:10.1073/pnas.0709518105. PMID 18364395. PMC 2278211 (freier Volltext).
- Folding@Home Crushes Exascale Barrier, Now Faster Than Dozens of Supercomputers - ExtremeTech. In: www.extremetech.com. Abgerufen am 13. Mai 2020.
- Folding@home crowdsourced computing project passes 1 million downloads amid coronavirus research. In: VentureBeat, 31. März 2020. Abgerufen am 13. Mai 2020.
- The coronavirus pandemic turned Folding@Home into an exaFLOP supercomputer (en-us). In: Ars Technica, 14. April 2020. Abgerufen am 13. Mai 2020.
- Liam Tung: CERN throws 10,000 CPU cores at Folding@home coronavirus simulation project (en). In: ZDNet. Abgerufen am 13. Mai 2020.
- Senior, A. W., Evans, R., Jumper, J., Kirkpatrick, J., Sifre, L., Green, T., ... & Penedones, H. (2020). Improved protein structure prediction using potentials from deep learning. Nature, 577(7792), 706–710.