Polycythaemia vera
Die Polycythaemia vera (Abk. PV, auch echte Polycythämie oder echte Polyzythämie, Polycythaemia rubra vera oder primäre idiopathische Polycythaemia rubra vera[1] genannt; synonym: Morbus Vaquez-Osler und Vaquez-Osler-Krankheit[2]) ist eine seltene myeloproliferative (die myeloische Blutbildung im Knochenmark betreffende) Erkrankung, bei der eine abnorme Vermehrung von roten Blutzellen (Erythrozyten) vorliegt, ohne dass hierfür ein physiologischer Stimulus existiert. Zu den Hauptsymptomen zählen eine vermehrte Blutviskosität bis hin zum Hyperviskositätssyndrom und damit zusammenhängende Durchblutungsstörungen. Auch Bluthochdruck kann auftreten. In der Regel verläuft die Erkrankung relativ gutartig und wird zunächst[3] durch Aderlässe behandelt. Selten geht eine PV in andere Erkrankungen über (sekundäre Osteomyelofibrose, akute myeloische Leukämie). Es kann von einer nahezu normalen Lebenserwartung ausgegangen werden, wenn die prinzipiell unheilbare Erkrankung frühzeitig erkannt und adäquat lebenslang behandelt wird. Andernfalls ist ein maligner Verlauf zu erwarten.[4]
| Klassifikation nach ICD-10 | |
|---|---|
| D45 | Polycythaemia vera ICD-O 9950/3 |
| ICD-10 online (WHO-Version 2019) | |
_London.jpg.webp)
Wortbedeutung
Die altgriechisch-lateinischen Bestandteile des Namens beschreiben das Hauptmerkmal der Krankheit:
- poly (πολύς) = viel
- cyt bzw. zyt (κύτος „Gefäß“) = Zelle
- haem(ie) (αἷμα) = Blut(krankheit)
- vera = wahr, echt
Begriffsdefinitionen
Die Polycythaemia vera, d. h. die „echte“ oder „wahre“ Polyzythämie, wird seit 1892 (Henri Vaquez)[5] von anderen Formen der Polyzythämie, den reaktiven oder sekundären Polyzythämien, abgegrenzt.
Reaktive Polyzythämien entstehen allgemein als Reaktion auf einen anderen Stimulus, in der Regel eine Sauerstoffminderversorgung (Hypoxämie), wie sie z. B. bei chronischen Lungenerkrankungen, exzessivem Nikotinkonsum, Schlafapnoe (Atemaussetzer) oder Aufenthalt in großer Höhe (niedrigerer Sauerstoffpartialdruck) auftreten kann. Der menschliche Körper reagiert in solchen Situationen mit der vermehrten Bildung von Erythropoetin („Epo“), was eine vermehrte rote Blutbildung bewirkt. Menschen, die lange Zeit in großer Höhe leben (z. B. im Andenhochland von Bolivien), haben deswegen einen durchschnittlich höheren Hämoglobinwert und höhere Werte für Erythrozyten; sie haben eine „physiologische“ Polyzythämie, die nicht als Krankheit zählt. Sekundäre Polyzythämien können z. B. bei Erythropoetin-produzierenden Tumoren entstehen (sehr selten).
Die Polyzythaemia vera ist dagegen eine Krankheit mit einer genetischen Ursache, entstanden durch eine im Laufe des Lebens zufällig erworbene genetische Störung in hämatopoetischen Stammzellen,[6] dennoch werden familiäre Häufungen beobachtet.[7]
Epidemiologie
Die Prävalenz (Bevölkerungsanteil der Kranken) liegt in den USA bei 1:3.300 (≈ 0,03 %), die Inzidenz (jährliche Neuerkrankungsrate) bei 1:36.000 bis 1:100.000,[8] nach anderer Quelle[9] bei 1–2:100.000. Sie ist somit die häufigste Form der myeloproliferativen Erkrankungen. Da es für Deutschland keine epidemiologischen Daten gibt, wurde die schwedische Statistik herangezogen und daraus jährlich 2.000 Neuerkrankungen in Deutschland geschätzt.[10] Das Selbsthilfeforum mpn-Netzwerk spricht von 500 bis 600 Neuerkrankungen pro Jahr.[11] Sie kann in jeder Altersstufe auftreten, der Altersgipfel liegt zwischen der 5. und 6. Lebensdekade. Das Verhältnis zwischen männlichen und weiblichen Patienten beträgt etwa 2:1. Eine familiäre Häufung (die auf eine Vererbung schließen lässt) kommt vor, ist aber selten.[12]
Verlauf
Oftmals zeigen sich schon bis zu einem Jahrzehnt vor Eintritt der Polycythaemia vera in die chronische Phase einzelne latente Anzeichen der Krankheit, beispielsweise in Form einer mäßigen Vergrößerung der Blut-abbauenden Organe Milz und Leber. Das Blut-bildende Knochenmark hingegen weist keine erkennbaren Veränderungen auf. Die JAK2-Mutation (s. u.) ist jedoch schon nachweisbar.
Generell werden zwei Stadien der Polycythaemia vera unterschieden. Eine erst chronische Phase mit erhöhter Produktion der Erythrozyten kann zwei Jahrzehnte oder mehr bestehen, eine progrediente Spätphase mit diversen, auch akut lebensgefährdenden, Komplikationen kann bei einem Teil der Patienten daran anschließen.[13]
Ursachen und Entstehung
Im Jahr 2005 wurde durch mehrere wissenschaftliche Arbeitsgruppen eine Mutation im JAK2-Gen („Januskinase“ 2, eine Tyrosinkinase) beschrieben.[14][15] Diese Mutation der genomischen DNA führt zu einem Aminosäure-Austausch (Valin gegen Phenylalanin) an Position 617 des JAK2-Proteins („V617F-Mutation“). Das JAK2-Protein spielt eine wichtige Rolle bei der Signaltransduktion in der Zelle. Durch die Mutation wird es aktiviert, so dass betroffene Zellen dauerhaft eine gesteigerte Zellteilungsrate haben. Die V617F-Mutation findet man bei verschiedenen hämatologischen Erkrankungen, aber besonders häufig (in mehr als 90 %) bei der Polycythaemia vera. Die betroffenen blutbildenden Stammzellen sind von der Stimulation durch Erythropoetin (Epo) unabhängig und zeigen eine hundertfach erhöhte Sensitivität auf Wachstumsfaktoren wie IGF-1 (Insuline-like Growth Factor 1) und IL-3 (Interleukin-3).
Diagnosestellung
In den meisten Fällen wird die Erkrankung dadurch entdeckt, dass bei einem Blutbild – oft im Nebenbefund – eine massive Erhöhung des Hämatokrits, der Erythrozyten oder der Hämoglobinkonzentration auffällt. Seltener führen eher unspezifische Symptome wie Juckreiz, eine leichte Milzvergrößerung, Thrombosen, Tinnitus oder andere klinische Symptome dazu, dass anhand dieser Erstsymptome (Leitsymptome) gezielt nach der Polycythaemia vera gesucht wird.[13]
Diagnosekriterien der WHO
Die Vermehrung der Erythrozyten lässt sich im Labor durch Messung des Hämatokritwertes oder des Hämoglobins sowie des Erythropoetins nachweisen. Auch Leukozyten und Thrombozyten sind im Blutbild meist vermehrt. Die Blutsenkung (BSG) ist dagegen verlangsamt, häufig sind Harnsäure und LDH im Serum erhöht. Die Weltgesundheitsorganisation (WHO) hat die in der folgenden Tabelle zusammengestellten Diagnosekriterien aufgestellt.[16]
| Kriterium | Bedingungen |
|---|---|
| Major 1 | Hämoglobin > 16,5 g/dL bzw. >16,0 g/dL bei Männern/Frauen oder
Hämatokrit >49 % bzw. 48 % bei Männern/Frauen oder erhöhte Erythrozytenmenge (>25 % über dem mittleren Normalwert) |
| Major 2 | Knochenmarkbiopsie zeigt gemessen am Patientenalter Hyperzellularität mit Steigerung aller drei Reihen |
| Major 3 | Nachweis der JAK2 V617F-Mutation oder Nachweis der JAK2 Exon 12-Mutation |
| Minor 1 | Erythropoietin-Spiegel im Serum unterhalb des Normalbereichs |
Die Diagnose einer Polycythaemia vera kann gestellt werden,
wenn entweder:
- alle drei Major-Kriterien erfüllt sind,
oder:
- die ersten beiden Major-Kriterien und das Minor-Kriterium erfüllt sind. Anmerkung: Im Falle einer anhaltenden Erythrozytose mit Hämoglobinwerten von >18,5 g/dL bei Männern (Hämatokrit, 55,5 %), bzw. >16,5 g/dL bei Frauen (Hämatokrit, 49,5 %) wird Major 2 Kriterium für die Diagnose nicht benötigt, falls Major 3 Kriterium und Minor 1 Kriterium positiv sind.
Spezifische und unspezifische klinische Symptome
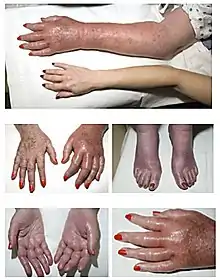
Durch die erhöhte Anzahl der Thrombozyten, Granulozyten und Erythrozyten wird das Blut dickflüssiger (Hyperviskosität). Es kann somit zu Durchblutungsstörungen in allen Bereichen des Körpers (insbesondere im Kapillarbereich) kommen, die Gefahr von Thrombosen und in der Folge von Embolien nimmt zu. Gleichzeitig kann auch die Blutungsgefahr steigen, weil die körpereigene Blutstillung als Ganzes nicht mehr einwandfrei abläuft.
Viele der möglichen Symptome der Polycythaemia vera sind nicht spezifisch für die Erkrankung, geben während der Anamnese und während des Krankheitsverlaufs den Ärzten und Patienten wichtige Hinweise. Zu möglichen (nicht bei allen Patienten auch feststellbaren) klinischen Symptomen zählen:[2][13]
- Splenomegalie (Vergrößerung der Milz)
- Hepatomegalie (Vergrößerung der Leber)
- Kopfschmerz
- Tinnitus (Ohrensausen)
- Sehstörungen
- Vertigo (Schwindel)
- Müdigkeit
- dysphorische (misslaunische) oder depressive Stimmungslagen
- Parästhesien
- Kribbeln oder Taubheitsgefühle insbesondere an den Extremitäten
- Schmerzen in den Extremitäten
- Pruritus (Juckreiz)
- besonders bei Wärme
- aquagener Pruritus (stechender Juckreiz nach Kontakt mit Wasser)
- Dyspnoe (Atemnot)
- Plethora
- Haut und Schleimhäute tiefrot mit rotblauer Zyanose
- rotes Gesicht – Patienten haben ein „blühendes“ Aussehen
- bei einer Augenuntersuchung oft dunkel-rot-bläulich gefärbte Gefäße
- Durchblutungsstörungen
- arterielle und venöse Thrombosen und/oder Embolien vor allem im Magen- und Darmbereich (Gastrointestinaltrakt)
- transitorische ischämische Attacken
- Magen- und Darmgeschwüre
- Arterielle Hypertonie (Bluthochdruck)
- gesteigerte Blutungsneigung, meist durch Funktionsstörungen der Thrombozyten (trotz erhöhter Anzahl)[17]
- selten Freisetzung erhöhter Mengen an Harnsäure bei vermehrtem Zerfall von Zellkernen der sich autonom vermehrenden Zellen mit Gichtanfällen[17]
- Nachtschweiß[18]
Bei fortschreitender Erkrankung können die Zahl der Erythrozyten und auch der Leukozyten und Thrombozyten sinken, die Milz kann an Größe zunehmen, oft in Verbindung mit Myelofibrose und extramedullärer Hämatopoese (Blutbildung außerhalb des Knochenmarks).
Ein Teil der individuell unterschiedlich auftretenden Symptome, insbesondere wenn sie mit Durchblutungsstörungen zusammenhängen, verschwindet oder wird abgeschwächt, sobald die Erkrankung erkannt und behandelt wird, indem der Hämatokritwert in den Normbereich von etwa 45 Prozent abgesenkt wird.
Differentialdiagnose
Sofern bei Patienten sowohl die Anzahl der Erythrozyten als auch die Anzahl der Leukozyten und Thrombozyten erhöht ist, ist die Diagnose einer Polycythaemia vera sehr wahrscheinlich. Sind lediglich die Erythrozyten vermehrt, muss ein umfangreiches differentialdiagnostisches Untersuchungsprogramm durchgeführt werden. Insbesondere ist abzuklären, ob als Ursache eventuell eine Exsikkose, eine Hypoxie, eine Kohlenmonoxidintoxikation, eine Erkrankung des Herzens (z. B. ein Rechts-links-Shunt), ein Hyperspleniesyndrom, paraneoplastische Syndrome, myelodysplastische Syndrome, eine Osteomyelofibrose, neurologische Erkrankungen oder auch Medikamente (z. B. Androgene oder Erythropoetin) ursächlich in Frage kommen.[13][12]
Therapie
Da sich eine endgültige Diagnose über mehrere Wochen hinziehen kann (z. B. bei der genetischen Bestimmung von JAK2), müssen unter Umständen schon während der Abklärung prophylaktisch geeignete Maßnahmen ergriffen werden, um einen lebensgefährlich hohen Hämatokritwert abzusenken. Im Wesentlichen verläuft eine Therapie auf mehreren Schienen:[13][6]
- Senkung des Hämatokritwerts durch regelmäßige Aderlässe bzw. Apherese,
- eventuelle Thrombozytenaggregationshemmung zur Verminderung der erhöhten Thrombosegefahr durch Acetylsalicylsäure (z. B. Aspirin) oder Antikoagulanzien (sofern die Blutgerinnung nicht krankheitsbedingt gestört ist)
- Zellreduktive Medikamente (Hydroxycarbamid) ("milde Form der Chemotherapie"), falls die anderen Maßnahmen nicht mehr ausreichen oder die Anzahl der Thrombozyten in einem Bereich liegt, der die Thrombosegefahr gefährlich erhöht
- Alpha-Interferontherapie mit Ropeginterferon als Alternative zur Chemotherapie
- Ruxolitinib bzw. INC 424 (Handelsname Jakavi des Pharmaunternehmens Novartis), wie auch Givinostat (2017 noch in der Studienphase, inzwischen in der Zweitlinientherapie zugelassen[19]) ein Januskinase-Inhibitor (-Hemmer), zugelassen seit 2015 bei Resistenz od. Intoleranz gegenüber Hydroxycarbamid oder bei starker Milzvergrößerung und zur Behandlung der Post-Polycythaemia-vera-Myelofibrose.[20]
Sonstige, die Erkrankung individuell begleitende Beschwerden, können nur symptomatisch behandelt werden. Ruxolitinib wirkt jedoch in vielen Fällen auch positiv auf die sogenannten Sekundärsymptome (z. B. Juckreiz, Milzvergrößerung, Müdigkeit).
Aderlass und Apherese
Als wichtigste Maßnahme dienen bei Diagnosestellung häufige (wöchentliche), später (nach Erreichen der Normalwerte) regelmäßige (6 bis 10 Wochen) Aderlässe zur Reduktion der Erythrozyten und in geringerem Maße anderer zellulärer Bestandteile des Blutes.

Als alternative Maßnahme zu mehrfachen Aderlässen findet die therapeutische Apherese (bekannt als Blutwäsche) Anwendung. Dieses Verfahren filtert die überzähligen Blutbestandteile in einem etwa 20-minütigen Arbeitsgang heraus. Während ein Aderlass den Hämatokrit um maximal 3 Prozent absenkt, lässt er sich bei einer Apherese gezielt um bis 12 Prozent reduzieren. Bei anschließenden gelegentlichen Blutwertkontrollen ist eine erneute Anwendung in drei- bis sechsmonatigem Abstand erforderlich. Derzeit gehört die Apherese nicht zur Standardtherapie und muss ärztlicherseits begründet werden. Wegen der aufwändigen Technik und des damit verbundenen Kostenaufwands wird eine Apherese bundesweit in wenigen Krankenhäusern, kaum in hämatologischen Praxen, durchgeführt.
Häufig steigt während der Behandlung mittels Aderlässen (Phlebotomie) oder Blutwäschen (Apherese) der Anteil der Thrombozyten und der Leukozyten, da durch diese Maßnahmen nur der Anteil der roten Blutkörperchen mittelfristig gesenkt werden kann. Die anderen festen Blutbestandteile (Thrombo- und Leukozyten) werden krankheitsbedingt weiterhin – individuell unterschiedlich – im erhöhten Maße produziert. Nach Absenken des Hämatokrits auf etwa 45 Prozent muss insbesondere der Thrombozytenanteil beobachtet werden.
Thrombozytenaggregationshemmung
Durch die erhöhte Gefahr, dass sich Blutzellen, insbesondere Thrombozyten (Thrombozytenaggregation) verklumpen und Blutgefäße verstopfen (Thrombosen) und dass sich diese Thrombosen lösen und an einer anderen Stelle des Blutkreislaufs den Blutfluss blockieren (Embolie), ist es wichtig, die erhöhte Thromboseneigung zu vermindern. Ursache dieser Neigung zur Thromboembolie sind einerseits die erhöhte Anzahl an Blutzellen (besonders bei stark erhöhten Werten an Erythrozyten[13] und Thrombozyten) und andererseits eine noch nicht gänzlich verstandene Störung des gesamten Regelmechanismus der Blutgerinnung. Bei Vorliegen einer JAK2-V617F Mutation ist die Thromboseneigung zudem durch eine erhöhte Anlagerungsneigung der Granulozyten an den Gefäßwänden erhöht.[21]
Sofern es keine Kontraindikationen (Gegenanzeigen) gibt, wie z. B. eine erhöhte Blutungsneigung, kann durch dauerhafte Einnahme von oralen Thrombozytenaggregationshemmern wie Acetylsalicylsäure (ASS) in relativ niedriger Dosierung (50 bzw. 100 mg pro Tag) das Thromboserisiko vermindert werden.[22] Die Einnahme erfolgt im Allgemeinen schon vor Beginn und parallel zur Aderlasstherapie. In einem Teil der Fachliteratur wird kritisch darauf hingewiesen, derartige Medikamente wegen möglicher Nebenwirkungen nur dann einzunehmen, wenn bereits Thrombosen erfolgt sind und dass gleichzeitig die Erythrozytenmasse strikt zu kontrollieren sei.[13]
Zellreduktive-, Interferontherapie
Solange die notwendige Absenkung des Hämatokrits durch Aderlässe erreicht werden kann und keine thromboembolischen Komplikationen auftreten, ist eine Chemotherapie nicht indiziert. Falls eine zu hohe Anzahl an Leukozyten oder Thrombozyten vermehrt zu Thrombosen und/oder Embolien führt, falls eine sehr starke Milzvergrößerung feststellbar ist und/oder ein nach Aderlässen auftretender wasserinduzierter Juckreiz entsteht, ist zu untersuchen, ob eine Chemotherapie als zytoreduktive Maßnahme (Verminderung der zu hohen Bildung neuer Zellen) sinnvoll sein könnte. Ziel wäre es, die Neubildung insbesondere der Thrombozyten einzuschränken.[6]
Aktuell werden zur Behandlung der in Deutschland zugelassene Hydroxyharnstoff (z. B. Litalir), Ruxolitinib, Alpha-Interferon und in Erprobung Anagrelid eingesetzt. Wie bei jeder Chemotherapie sind Nebenwirkungen nicht auszuschließen. Als Nebenwirkungen können beispielsweise Schleimhautirritationen, Fieber, psychische Veränderungen, Hauttumore, starke Schwankungen der Thrombozytenwerte oder Durchfälle auftreten. In seltenen Fällen kann auch eine akute Leukämie ausgelöst werden.[6]
Weitere Maßnahmen
Aufgrund möglicherweise mit der Erkrankung verbundener, individuell sehr unterschiedlicher Symptome werden eine Reihe von ergänzenden Maßnahmen durchgeführt, die in erster Linie die Begleiterscheinungen lindern sollen. Da hierbei nicht ursächlich eingewirkt werden kann, hängt der Einsatz letztlich von der unmittelbaren Wirkung auf den jeweiligen Patienten ab.
Ergänzend kann die Gabe von Allopurinol wegen eines erhöhten Harnsäureanfalls (besonders während der zytoreduktiven Therapie) zur Prophylaxe eines Gichtanfalls oder einer Uratnephropathie angebracht (indiziert) sein. Im Allgemeinen ist es nicht notwendig, eine asymptomatische Hyperurikämie unter 10 mg/dl zu behandeln.[13]
Ein bei über der Hälfte der Patienten auftretender aquagener Juckreiz, der die Lebensqualität erheblich beeinträchtigen kann, kann ergänzend zu Aderlässen nur symptomatisch durch Badezusätze (z. B. Bicarbonat, Stärke), Antihistaminika, Serotonin-Wiederaufnahmehemmer, juckreizmindernde Cremes (z. B. mit Capsaicin) oder eine Phototherapie (nicht ungefährlich wegen einer möglichen Kanzerogenität) mehr oder weniger stark gelindert werden.[6]
Bei einzelnen Patienten mit extrem vergrößerter Milz, die sich medikamentös nicht mehr behandeln lässt, kann ein operativer Eingriff notwendig werden. Seit 2016 kann durch den Einsatz von Ruxolitinib in vielen Fällen eine rasche Verkleinerung der Milz bewirkt werden.
Bei jüngeren Patienten (je nach Gesundheitszustand, bis ca. 65 Jahre) wird im Extremfall und bei weit fortgeschrittener Erkrankung auch eine Knochenmark- bzw. Stammzellentransplantation erforderlich sein.[13]
Als Alternative bei älteren Patienten kann auch eine Radiophosphortherapie in Betracht gezogen werden.[23]
Prognose
Polycythaemia vera ist mit einem erhöhten Vorkommen an thromboembolischen Ereignissen, verschiedenen hämatologischen Komplikationen sowie einer reduzierten Lebenserwartung assoziiert.
Die durchschnittliche Lebenserwartung nach Krankheitsbeginn, bei unbehandelten Patienten mit Symptomen, wurde auf 18 Monate geschätzt.[24] Behandelte Patienten haben eine Lebenserwartung von mindestens 13 Jahren. Die Lebenserwartung behandelter Patienten ist dennoch geringer als die einer alters- und geschlechtsgleichen Normalpopulation.[25][26][27][28]
Die umfangreichste Analyse von Faktoren, die die Lebenserwartung bei Polycythaemia vera beeinflussen, war eine internationale Studie mit 1545 Patienten, die unterschiedliche Therapieformen erhielten. Die Studie identifizierte Alter, Leukozytose, Vorgeschichte venöser Thrombosen und einen abnormalen Karyotyp als unabhängige Risikofaktoren für die Lebenserwartung. Patienten unter 62 Jahren und mit einer Leukozytenzahl von ≤ 10.500 /µl hatten eine mediane Überlebenszeit (durchschnittliche Überlebenszeit nach Diagnosestellung) von 23 Jahren. Im Gegensatz dazu hatten Patienten, die keines oder nur eins der beiden Kriterien erfüllten, eine mediane Überlebenszeit von 9 Jahren.[25]
Thromboembolien sind die Hauptursache von Morbidität und Mortalität bei Polycythaemia vera.[29] Hohes Alter (65–70 Jahre) und bereits stattgefundene Thromboembolien sind die zwei Hauptvorhersageindikatoren für das thromboembolische Risiko bei Polycythaemia vera.[30][26][24][31] Die Beibehaltung des Hämatokrits unter 45 Prozent ist mit einer signifikant geringeren Rate thrombotischer Komplikationen verbunden.[32]
Literatur
- Tiziano Barbui, Jürgen Thiele u. a.: The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. In: Blood Cancer Journal. Band 8, Nr. 2, Februar 2018, doi:10.1038/s41408-018-0054-y (englisch).
- Jerry L. Spivak: Polycythaemia vera und andere myeloproliferative Erkrankungen. In: Manfred Dietel (Hrsg.): Harrisons Innere Medizin. Deutsche Ausgabe in Zusammenarbeit mit der Charité. 17. Auflage. Band 1, Teil 6. McGraw-Hill, Berlin 2009, ISBN 978-3-86541-310-9, Kap. 103, S. 838–844 (dt. Fassung Isrid Sturm und Bernd Dörken).
- F. P. Siegel, P. E. Petrides: Angeborene und erworbene Polyzythämien (Übersichtsarbeit). In: Deutsches Ärzteblatt. Band 105, Nr. 4, 2008, S. 62–68 (aerzteblatt.de [PDF]).
- Polycythaemia vera. In: Pschyrembel Klinisches Wörterbuch 2012. 263. Auflage. Walter de Gruyter, Berlin / Boston 2011, ISBN 978-3-11-025166-1, S. 1666.
Weblinks
- Eva Lengfelder, Gabriela M. Baerlocher, Konstanze Döhner, Heinz Gisslinger, Martin Grießhammer, Steffen Koschmieder, Petro E. Petrides: Polycythaemia Vera (PV). In: onkopedia.com. Deutsche Gesellschaft für Hämatologie und Medizinische Onkologie e.V., April 2019.
- Polycythaemia vera (PV): Häufig gestellte Fragen. In: mpn-netzwerk.de. mpn-netzwerk e. V. – Deutsche Leukämie- und Lymphomhilfe e. V., Oktober 2016.
Einzelnachweise
- Ludwig Heilmeyer, Herbert Begemann: Blut und Blutkrankheiten. In: Ludwig Heilmeyer (Hrsg.): Lehrbuch der Inneren Medizin. Springer-Verlag, Berlin/Göttingen/Heidelberg 1955; 2. Auflage ebenda 1961, S. 376–449, hier: S. 418.
- Polycythaemia vera. In: Pschyrembel Klinisches Wörterbuch 2012. 263. Auflage. Walter de Gruyter, Berlin/Boston 2011, ISBN 978-3-11-025166-1, S. 1666.
- Onkopedia: Kapitel 6: Therapie
- Seltene Erkrankungen: Polycythämia Vera
- Ludwig Heilmeyer, Herbert Begemann: Blut und Blutkrankheiten. In: Ludwig Heilmeyer (Hrsg.): Lehrbuch der Inneren Medizin. Springer-Verlag, Berlin/Göttingen/Heidelberg 1955; 2. Auflage ebenda 1961, S. 376–449, hier: S. 418 f.
- F. P. Siegel, P. E. Petrides: Angeborene und erworbene Polyzythämien (Übersichtsarbeit). In: Deutsches Ärzteblatt. Band 105, Nr. 4, 2008, S. 62–68 (aerzteblatt.de [PDF; abgerufen am 15. April 2012]).
- Minimed: Polycythämia Vera Definition
- Eintrag bei Orphanet, Stand 2010, abgerufen 4. Mai 2016.
- Wolfgang Gerok: Die Innere Medizin: Referenzwerk für den Facharzt. Schattauer Verlag, 2007, ISBN 978-3-7945-2222-4, S. 68.
- F. P. Siegel, P. E. Petrides: Angeborene und erworbene Polyzythämien. In: Deutsches Ärzteblatt. Band 105, Nr. 4, 2008, S. 62–68, doi:10.3238/arztebl.2008.0062
- Polycythaemia vera (PV): Häufig gestellte Fragen. MPN-Netzwerk e. V., August 2015 (abgerufen 4. Mai 2016)
- verschiedene Quellen, darunter das medizinische Portal med2click, abgerufen am 5. März 2012.
- Jerry L. Spivak: Polycythaemia vera und andere myeloproliferative Erkrankungen. In: Manfred Dietel (Hrsg.): Harrisons Innere Medizin. Deutsche Ausgabe in Zusammenarbeit mit der Charité. 17. Auflage. Band 1, Teil 6. McGraw-Hill Inc., Berlin 2009, ISBN 978-3-86541-310-9, Kap. 103, S. 838–844 (dt. Fassung Isrid Sturm und Bernd Dörken).
- R. Kralovics, F. Passamonti, AS Buser u. a.: A gain-of-function mutation of JAK2 in myeloproliferative disorders. In: N Engl J Med 352, 2005, S. 1779–1790. Abstrac
- C. James, V. Ugo, JP Le Couedic u. a.: A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. In: Nature, 2005, 434, S. 1144–1148
- D. A. Arber, A. Orazi, R. Hasserjian, J. Thiele, M. J. Borowitz, M. M. Le Beau, C. D. Bloomfield, M. Cazzola, J. W. Vardiman: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. In: Blood. 127, 2016, S. 2391–2406, doi:10.1182/blood-2016-03-643544.
- Classen, Meinhard., Berdel, Wolfgang E.: Innere Medizin: mit 1246 Tabellen, 216 Kasuistiken, 450 Zusammenfassungen und 183 Praxisfragen. Urban & Fischer, 2004, ISBN 3-437-42830-6.
- Häufige Fragen zur Polycythaemia vera. (Memento des Originals vom 5. Februar 2012 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. MPD-Netzwerk; abgerufen am 5. April 2012
- Klinische Studie zur Wirksamkeit bei Polycythaemia Vera Patienten Stand 2. September 2016.
- Rote Liste. Januar 2016.
- N. Gupta, B. Edelmann, T. M. Schnoeder, F. C. Saalfeld, D. Wolleschak, S. Kliche, B. Schraven, F. H. Heidel und T. Fischer: JAK2-V617F activates β1-integrin-mediated adhesion of granulocytes to vascular cell adhesion molecule 1. In: Leukemia 31, 2017, S. 1223–1226, doi:10.1038/leu.2017.26
- R. Landolfi, R. Marchioli, J. Kutti u. a.: Efficacy and safety of low-dose aspirin in polycythemia vera. In: The New England Journal of Medicine 350, 2004, S. 114–124, PMID 14711910.
- Leitlinie der Deutschen Gesellschaft für Hämatologie/Onkologie Stand Juni 2014, abgerufen am 25. Januar 2015.
- E. CHIEVITZ, T. THIEDE: Complications and causes of death in polycythaemia vera. In: Acta medica Scandinavica. Band 172, November 1962, S. 513–523, doi:10.1111/j.0954-6820.1962.tb07186.x, PMID 14020816.
- A. Tefferi, E. Rumi, G. Finazzi, H. Gisslinger, A M Vannucchi, F. Rodeghiero, M L Randi, R. Vaidya, M. Cazzola, A. Rambaldi, B. Gisslinger, L. Pieri, M. Ruggeri, I. Bertozzi, N H Sulai, I. Casetti, A. Carobbio, G. Jeryczynski, D R Larson, L. Müllauer, A. Pardanani, J. Thiele, F. Passamonti, T. Barbui: Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. In: Leukemia. 27, 2013, S. 1874, doi:10.1038/leu.2013.163.
- Francesco Passamonti, Elisa Rumi, Ester Pungolino, Lucia Malabarba, Paola Bertazzoni, Marina Valentini, Ester Orlandi, Luca Arcaini, Ercole Brusamolino, Cristiana Pascutto, Mario Cazzola, Enrica Morra, Mario Lazzarino: Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. In: The American Journal of Medicine. 117, 2004, S. 755, doi:10.1016/j.amjmed.2004.06.032.
- . Gruppo Italiano Studio Policitemia*: Polycythemia Vera: The Natural History of 1213 Patients Followed for 20 Years. In: Annals of Internal Medicine. 123, 1995, S. 656, doi:10.7326/0003-4819-123-9-199511010-00003.
- Ayalew Tefferi, Paola Guglielmelli, Dirk R. Larson, Christy Finke, Emnet A. Wassie, Lisa Pieri, Naseema Gangat, Rajmonda Fjerza, Alem A. Belachew, Terra L. Lasho, Rhett P. Ketterling, Curtis A. Hanson, Alessandro Rambaldi, Guido Finazzi, Juergen Thiele, Tiziano Barbui, Animesh Pardanani, Alessandro M. Vannucchi: Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. In: Blood. 124, 2014, S. 2507, doi:10.1182/blood-2014-05-579136.
- Tiziano Barbui, Alessandra Carobbio, Elisa Rumi, Guido Finazzi, Heinz Gisslinger, Francesco Rodeghiero, Maria Luigia Randi, Alessandro Rambaldi, Bettina Gisslinger, Lisa Pieri, Irene Bertozzi, Ilaria Casetti, Animesh Pardanani, Francesco Passamonti, Alessandro M. Vannucchi, Ayalew Tefferi: In contemporary patients with polycythemia vera, rates of thrombosis and risk factors delineate a new clinical epidemiology. In: Blood. 124, 2014, S. 3021, doi:10.1182/blood-2014-07-591610.
- Roberto Marchioli, Guido Finazzi, Raffaele Landolfi, Jack Kutti, Heinz Gisslinger, Carlo Patrono, Raphael Marilus, Ana Villegas, Gianni Tognoni, Tiziano Barbui: Vascular and Neoplastic Risk in a Large Cohort of Patients With Polycythemia Vera. In: Journal of Clinical Oncology. 23, 2005, S. 2224, doi:10.1200/JCO.2005.07.062.
- JOHN H. LAWRENCE, NATHANIEL I. BERLIN, REX L. HUFF: THE NATURE AND TREATMENT OF POLYCYTHEMIA STUDIES ON 263 PATIENTS. In: Medicine. 32, 1953, S. 323, doi:10.1097/00005792-195309000-00002.
- Roberto Marchioli, Guido Finazzi, Giorgina Specchia, Rossella Cacciola, Riccardo Cavazzina, Daniela Cilloni, Valerio De Stefano, Elena Elli, Alessandra Iurlo, Roberto Latagliata, Francesca Lunghi, Monia Lunghi, Rosa Maria Marfisi, Pellegrino Musto, Arianna Masciulli, Caterina Musolino, Nicola Cascavilla, Giovanni Quarta, Maria Luigia Randi, Davide Rapezzi, Marco Ruggeri, Elisa Rumi, Anna Rita Scortechini, Simone Santini, Marco Scarano, Sergio Siragusa, Antonio Spadea, Alessia Tieghi, Emanuele Angelucci, Giuseppe Visani, Alessandro Maria Vannucchi, Tiziano Barbui: Cardiovascular Events and Intensity of Treatment in Polycythemia Vera. In: New England Journal of Medicine. 368, 2013, S. 22, doi:10.1056/NEJMoa1208500.