Gastrointestinaler Stromatumor
Als gastrointestinaler Stromatumor (GIST) wird ein seltener bösartiger Bindegewebstumor (Sarkom) des Magen-Darm-Traktes (Gastrointestinaltrakt, von gr. Gaster „Magen“ und lat. Intestinum „Darm“) bezeichnet. Gastrointestinale Stromatumoren treten am häufigsten im Bereich des Magens auf und verursachen unspezifische Beschwerden. Grundlage der Behandlung ist üblicherweise die vollständige chirurgische Tumorentfernung, die unter Umständen durch eine medikamentöse Behandlung mit dem Tyrosinkinaseinhibitor Imatinib ergänzt wird. Die Prognose wird durch Größe, Lymphknotenbeteiligung, Metastasierungsgrad und Differenzierung bzw. der mitotische Aktivität des Tumors bestimmt.[1]
| Klassifikation nach ICD-10 | |
|---|---|
| C15.- | Bösartige Neubildung des Ösophagus |
| C16.- | Bösartige Neubildung des Magens |
| C17.- | Bösartige Neubildung des Dünndarms |
| C18.- | Bösartige Neubildung des Kolons |
| C19.- | Bösartige Neubildung des Retroperitoneums und des Peritoneums |
| ICD-10 online (WHO-Version 2019) | |
Häufigkeit
Die Inzidenz gastrointestinaler Stromatumoren wird in verschiedenen Studien mit zehn bis 20 Neuerkrankungen je einer Million Einwohner und Jahr angegeben. Männer sind geringfügig häufiger betroffen als Frauen. Das mediane Erkrankungsalter liegt bei 60 bis 70 Jahren. Eine US-amerikanische Studie zeigte eine etwas höhere Erkrankungswahrscheinlichkeit und auch Mortalität bei Afroamerikanern.[2][3]
Lokalisation
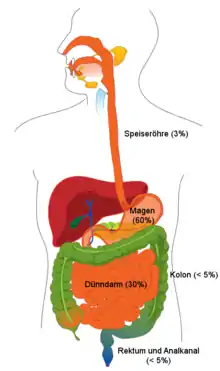
Der Magen ist mit etwa 60 % der häufigste Manifestationsort eines GIST, gefolgt von Dünndarm (30 %), Colon (< 5 %), Mastdarm und Analkanal (< 5 %) sowie Speiseröhre (3 %).[4]
Selten werden auch Tumoren beobachtet, die morphologisch und immunhistochemisch die Kriterien eines GIST erfüllen, aber keinen Bezug zur Wandung des Gastrointestinaltraktes zeigen. Solche Neoplasien werden auch als extragastrointestinale Stromatumoren (eGIST) bezeichnet und müssen von metastatischen Absiedlungen unterschieden werden. Bevorzugte Manifestationsorte von eGISTs sind Mesenterium, großes Netz[5] und Retroperitoneum. Einzelfallberichte dokumentieren das Auftreten von eGISTs an vielen weiteren Lokalisationen (unter anderem Bauchspeicheldrüse,[6] Gebärmutter,[7] Eileiter,[8] Ovarien,[9] Zwerchfell,[10] äußeres Genitale[11][12]).
Krankheitsursache und -entstehung
Die Krankheitsentität wurde erstmals 1983 auf der Grundlage neuer immunhistochemischer Erkenntnisse beschrieben, nachdem GISTs wegen ihrer lichtmikroskopischen Ähnlichkeit mit glattmuskulären oder neuralen Neoplasien früher häufig als Leiomyosarkome, Leiomyome, Leiomyoblastome oder Schwannome eingeordnet worden waren.[15] Neuere Forschungsergebnisse deuten auf eine Abstammung des Tumors von pluripotenten mesenchymalen Stammzellen hin, die Eigenschaften der interstitiellen Cajal-Zellen zeigen.[16] Bei letzteren handelt es sich um in der muskulären Wandung des Magen-Darm-Traktes lokalisierte Zellen mesenchymaler Herkunft, welche sowohl Merkmale glatter Muskulatur wie auch autonomer Nervenzellen zeigen und denen eine Schrittmacherfunktion im Rahmen der gastrointestinalen Peristaltik zukommt.
Ein maßgeblicher Beitrag zur Abgrenzung der gastrointestinalen Stromatumoren als eigenständige Tumorentität war 1998 der Nachweis von Mutationen des c-Kit-Protoonkogens in diesen Tumoren.[3][17]
Wesentliche Bedingung für die Entstehung der meisten gastrointestinalen Stromatumoren sind gain-of-function (GOF) Mutationen, die zur Bildung konstitutiv aktivierter Wachstumsfaktor-Rezeptoren an der Zelloberfläche führen. Diese entstehen meistens somatisch als Neumutation, kommen aber in seltenen Fällen auch bereits in der Keimbahn vor und sind dann erblich.
Sie betreffen meist den KIT-Rezeptor (ca. 90 %), sowie in einer Minderzahl der Fälle (ca. 5 %) den PDGFA (Platelet derived growth factor A)-Rezeptor. Normalerweise werden diese Rezeptoren erst nach Bindung eines spezifischen Moleküls (Ligand), im Falle des KIT-Rezeptors auch Stammzellfaktor genannt, aktiviert und vermitteln daraufhin ein Signal, das Zellwachstum und -proliferation fördert. Bei bestimmten, durch Mutationen bedingten Strukturänderungen dieser Rezeptoren kommt es jedoch zu deren permanenter Aktivierung auch in Abwesenheit eines Liganden, so dass das Gleichgewicht zwischen Zellwachstum und -proliferation einerseits und Zelltod (Apoptose) andererseits in Richtung der ersteren verschoben wird und ein abnormes Zellwachstum und eine Tumorbildung resultiert.
Bei einem geringen Anteil gastrointestinaler Stromatumoren werden keine bekannten Mutationen gefunden. Man spricht hier vom Wildtyp des Tumors; die an der Tumorentstehung beteiligten Mechanismen bleiben in diesen Fällen unverstanden.[3]
Assoziation mit anderen Erkrankungen und familiäre Formen
In einem kleinen Anteil der Fälle kann eine Beziehung zu erblichen Erkrankungen bestehen.
Beim seltenen familiären GIST-Syndrom liegen konstitutiv aktivierende Mutationen in den Genen KIT oder PDGFA bereits in der Keimbahn der Betroffenen vor, d. h. auch in deren Keimzellen (Spermien bzw. Eizellen), so dass sie auf Kinder weitervererbt werden können. In den bislang beobachteten Fällen lag dabei meist ein autosomal-dominanter Vererbungsmodus vor, so dass Nachkommen eines Betroffenen mit einer 50%igen Wahrscheinlichkeit ebenfalls zu Trägern des defekten Genes werden. Familiäre c-KIT-Mutationen haben dabei neben einer erhöhten Inzidenz von GISTs offenbar eine variable Begleitsymptomatik (verstärkte Hautpigmentierung, Nävi, Urticaria pigmentosa) zur Folge.[18]
Im Falle des seltenen, gleichfalls autosomal-dominant erblichen Carney-Stratakis-Syndroms wird eine Assoziation mit GISTs und Paragangliomen beobachtet. Ursächlich sind hier Mutationen von Genen, die für Untereinheiten der Succinatdehydrogenase (SDH) kodieren.[19][20]
Zusätzlich wird eine erhöhte Inzidenz von GISTs bei Patienten mit einer Neurofibromatose Typ 1, die Mutationen im NF1-Gen aufweisen, beobachtet.
Da bei allen diesen erblichen Formen die ursächliche Mutation bereits bei Geburt und in allen Körperzellen vorliegt, manifestieren sich familiäre GIST-Formen häufiger im jüngeren Lebensalter und zuweilen multizentrisch. In diesem Zusammenhang wurden Cajal-Zell-Hyperplasien als mögliche Vorläuferläsionen des GIST beschrieben.[18] Daneben gibt es Hinweise darauf, dass GIST-Patienten in vermehrtem Umfang gleichzeitig an weiteren malignen Tumoren erkranken (z. B. Adenokarzinome von Dickdarm und Magen, Lymphome des Magens).[21][22]
Bei der nicht erblichen Carney-Trias wird eine Assoziation von GISTs, Paragangliomen und Chondromen der Lunge beobachtet.[23]
Symptome
Die durch GIST verursachten Symptome sind unspezifisch. Zum Zeitpunkt der Diagnosestellung bestehen bei etwa 75 % der Patienten Beschwerden. Hierzu zählen Bauchschmerz (36 %), gastrointestinale Blutungen (25 %), eine Verschlechterung des Allgemeinzustandes (24 %), Verdauungsstörungen (16 %), eine Eisenmangelanämie (15 %), Übelkeit und Erbrechen (12 %) sowie Verstopfung oder Durchfall (9 %). Ein Tumor im Bauch ist nur bei etwa 8 % der Patienten tastbar.[24]
Diagnostik
Es gibt derzeit keine wegweisenden laborchemischen Untersuchungen, durch die sich die Diagnose eines GIST erhärten oder ausschließen ließe. Auch bildet der Tumor nach derzeitigem Kenntnisstand keine Faktoren, die im Blut nachweisbar und zur Frühdiagnose im Rahmen eines Screenings nutzbar wären.
Die Mehrzahl der GISTs sind einer endoskopischen Untersuchung zugänglich (Ösophagogastroduodenoskopie, Koloskopie), wobei gegebenenfalls die Entnahme einer Biopsie möglich ist.
Einen wesentlichen Beitrag zur Tumordiagnose und Bestimmung der Tumorausbreitung liefern bildgebende Verfahren wie Sonografie, konventionelle Röntgenaufnahmen nach Verabreichung von Kontrastmitteln, die Computertomografie sowie die Magnetresonanztomografie. Die Positronenemissionstomografie (PET) unter Verwendung von 18F-2-Fluor-2-deoxy-D-glucose eignet sich speziell zur Erfassung von Metastasen.[3]
Die eigentliche Diagnosestellung erfolgt durch histologische Untersuchung des Operationspräparates oder einer Biopsie durch einen Pathologen. Große Bedeutung kommt hierbei immunhistochemischen Untersuchungen (c-kit (CD117), CD34, DOG1) zu, da nur sie mit hinreichender Sicherheit die Unterscheidung von einigen morphologisch sehr ähnlichen Differentialdiagnosen erlauben.[3]
Differentialdiagnose
Bei der Diagnosefindung zu bedenken sind unter anderem eine Reihe weiterer gutartiger (z. B. Leiomyom, Lipom, Schwannom) und bösartiger Tumoren (z. B. Leiomyosarkom, Adenokarzinom, Lymphom, neuroendokrine Tumoren) sowie auch nicht-neoplastische Läsionen des Gastrointestinaltraktes wie eine arteriovenöse Malformation oder ektopes Bauchspeicheldrüsengewebe.[3][25]
Pathologie
.jpg.webp)
_C-KIT.jpg.webp)
GISTs erscheinen makroskopisch als grau-weiße, verfestigte bis markig weiche, kugelige oder angedeutet gelappte Tumoren mit einer Größe von 0,5–40 cm. Die Tumorgröße beträgt bei symptomatischen Patienten zum Zeitpunkt der Diagnosestellung im Mittel ca. 6,0 cm.[3][26] Typischerweise findet sich der Tumor in der Wandung eines gastrointestinalen Hohlorgans, unterhalb der Schleimhaut (submukös) gelegen und diese vorwölbend. Die Schleimhautbedeckung erscheint dabei häufig intakt, kann aber auch geschwürartig aufgebrochen sein. Nekrosen und Blutungen innerhalb der Tumormasse kommen wie auch zum Teil ausgeprägte zystische Umwandlungen vor.
Feingeweblich zeigen GISTs entweder ein rein spindelzelliges (69 %), epitheloides (12 %) oder gemischt spindelzelliges/epitheloides (20 %) Bild.[27] Immunhistochemisch exprimieren die überwiegende Mehrheit der GISTs die Marker c-Kit (CD117; 87 %) und PDGFA (66 %).[28] In 30–80 % der Fälle lassen sich auch glattmuskuläre Marker nachweisen; der zuverlässigste von diesen, Desmin, wird jedoch nur von einer Minderzahl der GISTs exprimiert. Eine CD 34-Positivität kann in bis zu 70 % der Fälle demonstriert werden.[29]
Klassifikation
Mit der 7. Auflage der TNM-Klassifikation maligner Tumoren (gültig ab Januar 2010) liegen nun erstmals vereinheitlichte Kriterien zur klinischen und pathologischen Tumorklassifikation des GIST vor:[30]
T (Primärtumor)
| TX | Primärtumor kann nicht beurteilt werden |
| T0 | Kein Anhalt für Primärtumor |
| T1 | Tumor 2 cm oder weniger in größter Ausdehnung |
| T2 | Tumor mehr als 2 cm, aber nicht mehr als 5 cm in größter Ausdehnung |
| T3 | Tumor mehr als 5 cm, aber nicht mehr als 10 cm in größter Ausdehnung |
| T4 | Tumor mehr als 10 cm in größter Ausdehnung |
N (regionäre Lymphknoten)
| NX | Regionäre Lymphknoten können nicht beurteilt werden |
| N0 | Keine regionären Lymphknotenmetastasen |
| N1 | Regionäre Lymphknotenmetastasen |
M (Fernmetastasen)
| M0 | Keine Fernmetastasen |
| M1 | Fernmetastasen |
Behandlung
Grundlage der Behandlung ist üblicherweise die vollständige chirurgische Entfernung des Tumors, die bei kleineren Geschwülsten auch minimal-invasiv (laparoskopisch) durchgeführt werden kann. Da GISTs nur selten Lymphknotenmetastasen ausbilden, ist eine routinemäßige Entfernung der regionären Lymphknoten nach gängiger Lehrmeinung nicht erforderlich.
Ein wesentlicher Fortschritt in der Behandlung des GIST war die Einführung des Tyrosinkinase-Inhibitors Imatinib (Glivec®), einem in Tablettenform zu verabreichenden Medikament, für das in Studien Ansprechraten von über 50 % belegt werden konnten.[3] Aufgrund der systemischen (den ganzen Körper betreffenden) Wirksamkeit ist es zur Behandlung von GISTs geeignet, die bereits metastasiert haben. Darüber hinaus wird auch der adjuvante und neoadjuvante Einsatz dieser Substanz erprobt. Bei Tumoren, die unzureichend oder nicht auf diese Therapie ansprechen, kann eine Behandlung mit dem Multikinaseinhibitor Sunitinib (Sutent®) versucht werden. Problematisch ist neben einer möglichen primären (bereits bei Behandlungsbeginn bestehenden) die Entwicklung einer sekundären (sich unter der Therapie entwickelnden) Resistenz der Tumorzellen gegen die Wirkung der Kinaseinhibitoren. Mögliche Ursache hierfür ist die Entwicklung sekundärer Mutationen des KIT-Gens[31][32] oder auch eine rhabdomyosarkomatöse Entartung.[33] Bei Resistenz gegen Imatinib und Sunitinib kann eine Behandlung mit dem Tyrosinkinaseinhibitor Regorafenib (Stivarga®) versucht werden. Derzeit in Erprobung ist der Einsatz weiterer, zum Teil bereits für andere Indikationen zugelassener Substanzen wie zum Beispiel Sorafenib (Nexavar®),[34] Nilotinib (Tasigna®)[35] oder Dasatinib (Sprycel®).[36]
Die konventionelle zytotoxische Chemotherapie mit Substanzen wie Doxorubicin, Ifosfamid, Dacarbazin oder Temozolomid zeigte in Studien, die vor Einführung von Imatinib durchgeführt wurden, enttäuschende Ergebnisse hinsichtlich der Rate an Tumorverkleinerungen. Wissenschaftliche Untersuchungen zur Chemotherapie von GIST, die nach einer Therapie mit Imatinib und Sunitinib behandelt wurden, liegen bisher noch nicht vor, allerdings sind auch längere Krankheitsstabilisierungen durch klassische Chemotherapie in dieser Situation möglich. GISTs erweisen sich überdies als weitgehend resistent gegen eine Strahlentherapie, die deshalb vorwiegend unter palliativer Zielsetzung, etwa wenn ein chirurgisches Vorgehen nicht möglich ist, zum Einsatz kommt.[3][37]
Prognose
Gastrointestinale Stromatumoren – vor allem die unter 2 cm großen und langsam wachsenden (welche die überwiegenden Mehrzahl darstellen) – haben generell eine gute Prognose. Durch die heute oft laparoskopisch durchgeführte Operation ist in den genannten Fällen meistens eine Heilung zu erreichen. Dies sieht bei metastasierten Tumoren anders aus. Die Krankheits-spezifische 5-Jahres-Überlebensrate bei Patienten mit metastasierten GISTs liegt verschiedenen Studien zufolge zwischen 30 und 60 %, wobei große Unterschiede zwischen lokalisierten Tumoren (mediane Überlebenszeit von 5 Jahren) einerseits und metastasierenden oder rezidivierenden Tumoren andererseits (mediane Überlebenszeit von 10–20 Monaten) zu verzeichnen sind. Entsprechend zeigen Tumoren, die aus operationstechnischen Gründen nicht oder nicht vollständig entfernt werden können, eine schlechte Prognose.[3] Eine Tumorruptur (spontan oder im Rahmen der Operation) verschlechtert wegen der Möglichkeit einer Tumoraussaat innerhalb der Bauchhöhle die Heilungsaussichten ebenfalls.[38] Die gleiche Gefahr besteht, wenn der Tumor an die peritoneale Bedeckung (Serosa) des befallenen gastrointestinalen Hohlorgans heranwächst und diese durchbricht.[39]
Bedeutende und auch in größeren Studien validierte Faktoren bei der Abschätzung der Prognose sind insbesondere Tumorgröße und die Geschwindigkeit, mit der sich die Tumorzellen teilen (mitotische Aktivität). Letztere bestimmt der untersuchende Pathologe, indem er unter dem Mikroskop die Zahl der Mitosefiguren in 50 Gesichtsfeldern bei hoher Vergrößerung („high power fields“, HPF) ermittelt. Aus diesen Parametern entwickelte man ein Einteilungsschema, nach dem sich Tumoren mit sehr niedrigem, niedrigem, intermediärem und hohem Risiko aggressiven biologischen Verhaltens (Rezidiv, Metastasierung) unterscheiden lassen:[40]
| Risiko | Tumorgröße | Mitotische Aktivität |
|---|---|---|
| sehr niedrig | < 2 cm | < 5 / 50 HPF |
| niedrig | 2–5 cm | < 5 / 50 HPF |
| intermediär | < 5 cm | 6–10 / 50 HPF |
| 5–10 cm | < 5 / 50 HPF | |
| hoch | > 5 cm | > 5 / 50 HPF |
| > 10 cm | jede mitotische Aktivität |
Weiterhin scheint auch der Sitz des Tumors einen Einfluss auf die Prognose zu haben. So zeigen GISTs des Magens eine bessere Prognose als die anderer Lokalisationen.[3] Ein Schema, das diesen Umstand berücksichtigt, und das auf Daten aus der Nachbeobachtung von über 1900 GIST-Patienten beruht, unterteilt das Patientenkollektiv in 8 Prognosegruppen:[41]
| Parameter | %-Anteil der Patienten mit progressiver Erkrankung/ Metastasierungsrisiko | |||||
|---|---|---|---|---|---|---|
| Gruppe | Tumorgröße [cm] | Mitosen [50 HPF] | Magen | Duodenum | Jejunum, Ileum | Rektum |
| 1 | ≤ 2 | ≤ 5 | 0 % kein | 0 % kein | 0 % kein | 0 % kein |
| 2 | > 2 ≤ 5 | ≤ 5 | 1,9 % sehr niedrig | 8,3 % niedrig | 4,3 % niedrig | 8,5 % niedrig |
| 3a | > 5 ≤ 10 | ≤ 5 | 3,6 % niedrig | 34 % hoch | 24 % mäßig | 57 % hoch |
| 3b | > 10 | ≤ 5 | 12 % mäßig | 34 % hoch | 52 % hoch | 57 % hoch |
| 4 | ≤ 2 | > 5 | 0 % | ? | 50 % | 54 % |
| 5 | > 2 ≤ 5 | > 5 | 16 % mäßig | 50 % hoch | 73 % hoch | 52 % hoch |
| 6a | > 5 ≤ 10 | > 5 | 55 % hoch | 86 % hoch | 85 % hoch | 71 % hoch |
| 6b | > 10 | > 5 | 86 % hoch | 86 % hoch | 90 % hoch | 71 % hoch |
In der Literatur hinsichtlich ihrer diagnostischen Signifikanz und Praktikabilität unterschiedlich bewertete ungünstige Prognosefaktoren sind eine hohe Zelldichte innerhalb des Tumors (Zellularität), eine epitheloidzellige oder gemischt epitheloidzellige/spindelzellige Histologie und Zell- und Kernatypien der Tumorzellen.[38][42]
Genetische Prognosefaktoren
Spezifische Mutationen stellen nach verschiedenen Studien unabhängige Prognosefaktoren dar. So zeigen Patienten, bei denen ein bestimmter Abschnitt des KIT-Gens verändert ist (Mutation des Exon 9) durchschnittlich einen ungünstigeren Krankheitsverlauf als Patienten mit anderen Mutationen (häufig Mutation des Exon 11). Liegen keine erfassbaren Mutationen vor (genetischer Wildtyp des GIST), ist dies mit einer besonders schlechten Prognose behaftet.[43]
Einzelnachweise
- Fauci u. a. (Hrsg.): Harrisons Innere Medizin. 17. Auflage. ABW, Berlin 2009, ISBN 978-3-86541-310-9, S. 761 f.
- T. Tran u. a.: The epidemiology of malignant gastrointestinal stromal tumors: an analysis of 1,458 cases from 1992 to 2000. In: Am J Gastroenterol. 2005 Jan;100(1), S. 162–168. PMID 15654796
- S. Mukherjee u. a.: Gastrointestinal Stromal Tumors. 4. März 2008.
- M. Miettinen u. a.: Evaluation of malignancy and prognosis of gastrointestinal stromal tumors: a review. In: Hum Pathol. 2002;33(5), S. 478–483. PMID 12094372
- Llenas-Garcíaet u. a.: Primary extragastrointestinal stromal tumors in the omentum and mesentery: a clinicopathological and immunohistochemical study. In: Hepatogastroenterology. 2008;55(84), S. 1002–1005. PMID 18705316
- Showalter u. a.: Extra-gastrointestinal stromal tumor of the pancreas: case report and a review of the literature. In: Arch Surg. 2008;143(3), S. 305–308. PMID 18347279
- Peitsidis u. a.: Extragastrointestinal stromal tumor mimicking a uterine tumor. A rare clinical entity. In: Int J Gynecol Cancer. 2008;18(5), S. 1115–1118. PMID 17986244
- Foster u. a.: Reclassification of a tubal leiomyosarcoma as an eGIST by molecular evaluation of c-KIT. In: Gynecol Oncol. 2006;101(2), S. 363–366. PMID 16439005
- Belics u. a.: Large gastrointestinal stromal tumor presenting as an ovarian tumor. A case report. In: J Reprod Med. 2003;48(8), S. 655–658. PMID 12971151.
- Yeung u. a.: Malignant extra-gastrointestinal stromal tumour of diaphragm. In: ANZ J Surg. 2008 Oct;78(10), S. 923–924. PMID 18959651
- Kang u. a.: Extragastrointestinal stromal tumor presenting as a scrotal mass: an unusual case. In: Asian J Androl. 2007;9(2), S. 275–279. PMID 17334596
- Lam u. a.: Extragastrointestinal stromal tumors presenting as vulvovaginal/rectovaginal septal masses: a diagnostic pitfall. In: Int J Gynecol Pathol. 2006;25(3), S. 288–292. PMID 16810068
- Miettinen & Lasota: Gastrointestinal stromal tumors; review on morphology, molecular pathology, prognosis, and differential diagnosis. In: Arch Pathol Lab Med. 2006, 130, S. 1466–1478. PMID 17090188
- Agaimy & Wünsch: Lymph node metastasis in gastrointestinal stromal tumours (GIST) occurs preferentially in young patients < or = 40 years: an overview based on our case material and the literature. In: Langenbecks Arch Surg. 2009;394(2), S. 375–381. PMID 19104826
- Mazur & Clark: Gastric stromal tumors. Reappraisal of histogenesis. In: Am J Surg Pathol. 1983;7(6), S. 507–519. PMID 6625048
- Kindblom u. a.: Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. In: Am J Pathol. 1998;152(5), S. 1259–1269. PMID 9588894
- Hirota u. a.: Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. In: Science. 1998;279(5350), S. 577–580. PMID 9438854
- Antonescu: Gastrointestinal stromal tumor (GIST) pathogenesis, familial GIST, and animal models. In: Semin Diagn Pathol. 2006;23(2), S. 63–69. PMID 17193819
- Carney & Stratakis: Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. In: Am J Med Genet. 2002 1;108(2), S. 132–139. PMID 11857563
- Pasini u. a.: Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. In: Eur J Hum Genet. 2008;16(1), S. 79–88. PMID 17667967
- Ł. Liszka u. a.: Coexistence of gastrointestinal stromal tumors with other neoplasms. In: J Gastroenterol. 2007;42(8), S. 641–649. PMID 17701127.
- Agaimy u. a.: Occurrence of other malignancies in patients with gastrointestinal stromal tumors. In: Semin Diagn Pathol. 2006;23(2), S. 120–129.
- Carney u. a.: The triad of gastric leiomyosarcoma, functioning extra-adrenal paraganglioma and pulmonary chondroma. In: N Engl J Med. 1977;296(26), S. 1517–1518. PMID 865533
- Mucciarini u. a.: Incidence and clinicopathologic features of gastrointestinal stromal tumors. A population-based study. In: BMC Cancer. 2007;7, S. 230. PMID 18096058
- Choti & Hanly: Gastric Stromal Tumors. (30. Juni 2006); http://emedicine.medscape.com/article/278845-overview
- Nilsson u. a.: Gastrointestinal stromal tumors: The incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era. In: Cancer. 2005;103(4), S. 821–829. PMID 15648083
- Koay u. a.: Gastrointestinal stromal tumours (GISTs): a clinicopathological and molecular study of 66 cases. In: Pathology. 2005;37(1), S. 22–31. PMID 15875730
- Zheng u. a.: Analysis of mutation and expression of c-kit and PDGFR-alpha gene in gastrointestinal stromal tumor. In: Hepatogastroenterology. 2007;54(80), S. 2285–2290. PMID 18265649
- Miettinen u. a.: Gastrointestinal stromal tumors--value of CD34 antigen in their identification and separation from true leiomyomas and schwannomas. In: Am J Surg Pathol. 1995;19(2), S. 207–216. PMID 7530409
- L. H. Sobin, M. K. Gospodarowicz, C. Wittekind: TNM classification of malignant tumors. 7. Auflage. Blackwell Publishing, 2010.
- Song u. a.: Secondary C-kit mutation is a cause of acquired resistance to imatinib in gastrointestinal stromal tumor. In: Scand J Gastroenterol. 2008; 18, S. 1–4. PMID 19096980
- Cassier u. a.: Novel approaches to gastrointestinal stromal tumors resistant to imatinib and sunitinib. In: Curr Gastroenterol Rep. 2008;10(6), S. 555–561.PMID 19006610
- Liegl u. a.: Rhabdomyosarcomatous Differentiation in Gastrointestinal Stromal Tumors After Tyrosine Kinase Inhibitor Therapy: A Novel Form of Tumor Progression. In: Am J Surg Pathol. 2009;33(2), S. 218–226 Abstract
- Nimeiri u. a.: Activity of sorafenib (SOR) in patients (pts) with imatinib (IM) and sunitinib (SU)-resistant (RES) gastrointestinal tumors (GIST): A phase II trial of the University of Chicago Phase II Consortium. 2008 Gastrointestinal Cancers Symposium.
- Deremer u. a.: Nilotinib: A second-generation tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia. In: Clin Ther. 2008;30(11), S. 1956–1975. PMID 19108785
- Schittenhelm u. a.: Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. In: Cancer Res. 2006;66(1), S. 473–481. PMID 16397263
- Rubin u. a.: Gastrointestinal stromal tumour. In: The Lancet. 2007;369(9574), S. 1731–1741. PMID 17512858.
- Rutkowski u. a.: Risk criteria and prognostic factors for predicting recurrences after resection of primary gastrointestinal stromal tumor. Ann Surg Oncol. 2007;14(7), S. 2018–2027. PMID 17473953
- Vallböhmer u. a.: Serosal penetration is an important prognostic factor for gastrointestinal stromal tumors. In: Oncol Rep. 2008;20(4), S. 779–783. PMID 18813818
- Fletcher u. a.: Diagnosis of gastrointestinal stromal tumors: A consensus approach. In: Hum Pathol. 2002 May;33(5), S. 459–465. PMID 12094370
- M. Mietinnen, J. Lasota: Gastrointestinal stromal tumors: Pathology and prognosis at different sites. In: Semin Diagn Pathol. 2006 May;23(2), S. 70–83. PMID 17193820
- Fletcher: Diagnostic Histopathology of Tumors. 3. Auflage. Churchill Livingstone Elsevier, 2007, S. 356–359.
- Heinrich u. a.: Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. In: J Clin Oncol: 2003;21, S. 4342–4349 PMID 14645423