Synovialsarkom
Das Synovialsarkom oder Synovialom[1] ist ein bösartiger Tumor, der vom Bindegewebe ausgeht und zu den Weichteilsarkomen gehört. Mikroskopisch ähnelt das Gewebe der Synovialmembran, obwohl man davon ausgeht, dass es sich nicht davon ableitet, sondern aus primitiven mesenchymalen Zellen entsteht.
| Klassifikation nach ICD-10 | |
|---|---|
| C49 | Bösartige Neubildung sonstigen Bindegewebes und anderer Weichteilgewebe |
| ICD-10 online (WHO-Version 2019) | |
Epidemiologie
Insgesamt sind Synovialsarkome sehr selten. Unter Kindern und Jugendlichen stellen sie aber, nach dem Rhabdomyosarkom, die häufigste Sarkomart dar. Über alle Altersgruppen gerechnet machen sie etwa 8 % aller Weichteilsarkome aus. Der Erkrankungsgipfel liegt zwischen 20 und 30 Jahren. Männer sind etwas häufiger betroffen als Frauen. Grundsätzlich können Synovialsarkome überall im Körper auftreten, bevorzugt sind aber die untere Extremität und hier vor allem die Umgebung der großen Gelenke.
Histologie
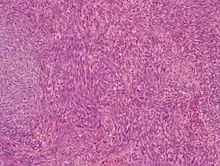
Es existieren zwei unterscheidbare gewebliche Typen: zum einen ein biphasisch genanntes Synovialsarkom, das aus drüsenartig angeordneten Epithelzellen in einem Spindelzellverband besteht, und dem monophysisch genannten Typus, der nur aus Spindelzellen aufgebaut ist.
Genetik
Synovialsarkome sind ganz überwiegend durch eine Translokation des X-Chromosoms und des Chromosoms 18 gekennzeichnet. Aus der Fusion zweier normaler Gene, zum einen SYT und zum anderen eines von zwei weitgehend homologen Genen, SSX1 oder SSX2, entsteht ein Fusionsgen, das dann abgelesen wird und ein pathologisches Fusionsprotein codiert. Auf welche Weise dies zur Entstehung des Tumors führt, ist noch nicht vollständig verstanden. Allerdings korreliert der Typ des Fusionsgens mit dem histologischen Subtyp und dem biologischen Verhalten des Sarkoms.[2]
Klinik
Die langsam wachsenden Tumoren sind meist gut abgegrenzt und enthalten oft zystische Anteile, weswegen sie leicht mit gutartigen Veränderungen verwechselt werden. Schmerzen fehlen zu Beginn meist, können aber im Verlauf durch die Kompression von umgebenden Strukturen entstehen. Zwischen den ersten Anzeichen und der Diagnosestellung können Monate, manchmal Jahre vergehen. Maßgeblich für die Diagnosestellung ist eine histologische Untersuchung. Die Größe des Tumors ist ein wichtiger Risikofaktor für eine Metastasierung, diese erfolgt meist in die Lungen. Die Behandlung erfolgt durch Operation, zusätzlich lokale Bestrahlung und gegebenenfalls Chemotherapie. Die Prognose ist günstig bei kleinen, nicht metastasierten Sarkomen mit hoher Zelldifferenzierung. Große, nicht komplett zu entfernende, und metastasierte Synovialsarkome gehen mit einer schlechten Prognose einher.
Weblinks
Einzelnachweise
- duden.de: Synovialom
- Kawai et al.: SYT–SSX Gene Fusion as a Determinant of Morphology and Prognosis in Synovial Sarcoma, N Engl J Med 1998; 338:153-160, doi:10.1056/NEJM199801153380303