Liposarkom
Das Liposarkom ist ein seltener bösartiger Tumor des Weichteilgewebes (Sarkom), der feingewebliche Merkmale von Fettzellen oder Fettzellvorstufen aufweist. Mit einem Anteil von 16–18 % ist das Liposarkom nach dem malignen fibrösen Histiozytom das zweithäufigste Weichteilsarkom.[1] Die Erstbeschreibung des Liposarkoms als Krankheitsentität erfolgte 1857 durch Rudolf Virchow.[2]
| Klassifikation nach ICD-10 | |
|---|---|
| C49.- | Bösartige Neubildung sonstigen Bindegewebes und anderer Weichteilgewebe |
| ICD-10 online (WHO-Version 2019) | |
| Klassifikation nach ICD-O-3 | |
|---|---|
| 8850/3 | Liposarkom o.n.A. |
| 8851/3 | Gut differenziertes Liposarkom |
| 8852/3 | Myxoides Liposarkom |
| 8853/3 | Rundzelliges Liposarkom |
| 8854/3 | Pleomorphes Liposarkom |
| 8855/3 | Gemischtzelliges Liposarkom |
| ICD-O-3 erste Revision online | |
Epidemiologie
Die Inzidenz des Liposarkoms wird international mit etwa 2,5 Neuerkrankungen je einer Million Einwohner und Jahr angegeben. Mit einem mittleren Erkrankungsalter von 50 Jahren handelt es sich um einen Tumor des Erwachsenen, der gleichwohl selten auch bei Kindern und jungen Erwachsenen beobachtet wird.[3][4] Männer sind geringfügig häufiger betroffen als Frauen. Geographische oder ethnische Häufigkeitsunterschiede wurden bislang nicht berichtet.[5]
Ätiologie
Die der Entstehung eines Liposarkoms zugrunde liegenden Ursachen sind weitgehend ungeklärt. Beschrieben wird eine mögliche Beziehung zu vorausgegangenen Verletzungen[6][7] und einer Exposition gegenüber ionisierender Strahlung.[8][9][10] Das Lipom, ein gutartiger und ungleich häufigerer Fettgewebstumor, ist keine typische Vorläuferveränderung des Liposarkoms, soll aber laut einigen Autoren in Einzelfällen dessen Ausgangspunkt bilden können. Andere Quellen bestreiten diese Ansicht und verweisen darauf, dass ein Übergang eines Lipoms in ein Liposarkom bislang nie überzeugend dokumentiert werden konnte.[11]
Pathologie
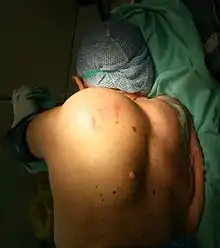
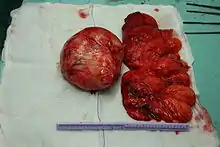
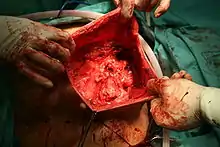
Vom makroskopischen Bild her sind Liposarkome oft relativ gut und häufig sogar kapselartig begrenzte, knotige oder gelappte, gelbliche bis grau-weiße Tumoren, die je nach Lokalisation eine erhebliche Größe und ein Gewicht von mehreren Kilogramm erreichen können. Die scheinbar gute Abgrenzung kann sich insofern als trügerisch erweisen, als in der Umgebung des Haupttumors zuweilen kleinere Tumorabsiedlungen gefunden werden. Liposarkome finden sich bevorzugt im tiefen Weichgewebe der unteren Extremität (59 %), der oberen Extremität (16 %), dem Retroperitoneum (15 %) und dem Körperstamm (8 %). Besonders häufig sind die Oberschenkel betroffen (41 %).[12]
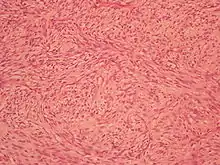
Durch die histologische (d. h. feingewebliche) Untersuchung lassen sich mehrere Subtypen des Liposarkoms unterschieden, die eine unterschiedliche Prognose zeigen und zum Teil auch bevorzugt in bestimmten Körperregionen auftreten:
| Histologischer Subtyp | Relative Häufigkeit | Dedifferenzierung | Bilder |
|---|---|---|---|
| Gut differenziertes Liposarkom | 40–45 % | niedriggradig | Makroskopie |
| Myxoides/rundzelliges Liposarkom | 30–35 % | mittelgradig/hochgradig | Makroskopie Histologie |
| Pleomorphes Liposarkom | 5 % | hochgradig | Makroskopie |
| Dedifferenziertes Liposarkom | selten | hochgradig | Makroskopie |
Der Grad der Dedifferenzierung eines Liposarkoms gibt an, wie stark sich das Tumorgewebe morphologisch vom reifen Fettgewebe unterscheidet. Dies ist deshalb bedeutsam, da mit zunehmender geweblicher Unreife auch ein zunehmend bösartigeres biologisches Verhalten des Tumors und eine schlechtere Prognose zu erwarten sind (aggressives lokales Wachstum, Rezidivneigung, Metastasierung). Für gut differenzierte Liposarkome werden zuweilen auch die Begriffe atypischer lipomatöser Tumor oder atypisches Lipom verwendet, da sie einerseits morphologisch einem Lipom sehr ähnlich sein können und zudem in Abwesenheit einer Tumorprogression nicht metastasieren, so dass ihnen ein prognostisch bedeutsames Merkmal maligner Tumoren fehlt. Es ist mittlerweile allgemein akzeptiert, dass rundzellige Liposarkome eine dedifferenzierte (geweblich unreifere und sich somit bösartiger verhaltende) Variante des myxoiden Liposarkoms darstellen.[13]
Molekularpathologie
Genetische Veränderungen sind häufig und betreffen unter anderem eine Region auf dem langen Arm des Chromosoms 12 (12q13-15) mit Amplifikation des MDM2-Gens (murine double minute oncogene) und des für die Cyclin-abhängige Kinase 4 codierenden Gens CDK4. Die damit einhergehende Überexpression der entsprechenden Gene kann auf RNA- und Proteinebene nachgewiesen werden und unter Umständen zur Abgrenzung sowohl gegenüber gutartigen Lipomen als auch anderen Weichteilsarkomen beitragen.[14][15]
Klinische Symptomatik
Liposarkome werden häufig erst in fortgeschritteneren Stadien als tief gelegene, langsam wachsende tumoröse Gewebsmasse klinisch auffällig. Die genaue Symptomatik wird dabei vorwiegend von der Lokalisation des Tumors bestimmt. Mit dem Tumorwachstum möglicherweise einhergehende Allgemeinerscheinungen sind zum Beispiel Müdigkeit, Abgeschlagenheit, Gewichtsverlust, Übelkeit und Erbrechen.
Diagnose
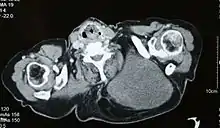
Bildgebende Verfahren wie die Computertomographie, die Magnetresonanztomographie, die Angiographie oder die Szintigraphie liefern diagnostische Hinweise und ermöglichen eine Einschätzung der Ausbreitung des Tumorleidens. Zur definitiven Diagnosesicherung ist in der Regel eine Biopsie und die histologische Untersuchung des gewonnenen Tumorgewebes durch einen Pathologen erforderlich.
Therapie
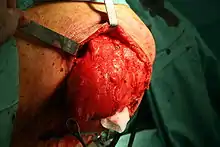
Der erfolgversprechendste therapeutische Ansatz ist die vollständige chirurgische Entfernung des Tumors unter Einhaltung eines ausreichenden Sicherheitsabstandes. Weitere Therapieoptionen sind die lokale Bestrahlung und eine Chemotherapie. Obwohl das Liposarkom als das strahlensensibelste Sarkom gilt, konnte eine Steigerung der Überlebenszeit durch eine Radiotherapie in wissenschaftlichen Studien bislang nicht überzeugend gezeigt werden. Auch die Chemotherapie des Liposarkoms hat gegenwärtig noch experimentellen Charakter.[16][17]
Prognose
Die Heilungsaussichten sind neben der Möglichkeit einer kompletten chirurgischen Entfernung davon abhängig, welcher feingewebliche Subtyp des Liposarkoms vorliegt. Die gut differenzierten sowie die meisten myxoiden Liposarkome zeigen mit einer Fünf-Jahres-Überlebensrate von 100 beziehungsweise 88 Prozent eine günstige Prognose. Diese ergibt sich unter anderem daraus, dass diese Formen kaum zu Metastasenbildung neigen. Hingegen versterben etwa 50 Prozent der Patienten mit einem rundzelligen oder schlecht differenzierten Liposarkom binnen fünf Jahren an ihrem Tumorleiden.[18] Metastatische Tumorabsiedlungen betreffen vor allem die Lunge (20 %), Knochen (8 %), Lymphknoten (6 %) und die Leber (5 %).[12]
Literatur
- A. N. Khan u. a.: Liposarcoma, Soft Tissue. (12. März 2008);
- R. A. Schwartz u. a.: Liposarcoma. (18. April 2008);
Einzelnachweise
- F. M. Enzinger und S. W. Weiss: Soft tissue tumours. 2. Ausgabe, St Louis, MO: Mosby-Year Book, 1998, S. 346–382.
- R. Virchow: Ein Fall von bösartigen zum Theil in der Form des Neuroms auftretenden Fettgeschwulsten. In: Virchows Arch A Pathol Anat Histopathol 11, 1857, S. 281–288.
- Z. Ahmed u. a.: Pleomorphic liposarcoma in a ten year old child. In: J Pak Med Assoc 54, 2004, S. 533–534. PMID 15552292
- E. Vocks u. a.: Myxoid liposarcoma in a 12-year-old girl. In: Pediatric Dermatology 17, 2000, S. 129–132. PMID 10792803
- R. A. Schwartz u. a.: Liposarcoma: Overview. Vom 16. Juli 2009
- S. D. Newlands u. a.: Mixed myxoid/round cell liposarcoma of the scalp. In: Am J Otolaryngol 24, 2003, S. 121–127. PMID 12649828 (Review)
- Nishimoto u. a.: A rare case of burn scar malignancy. In: Burns 22, 1996, S. 497–499. PMID 8884015
- H. Ninomiya u. a.: Postradiation sarcoma of the chest wall: report of two cases. In: Surg Today 36, 2006, S. 1101–1104. PMID 17123140
- D. Demir u. a.: Radiation-induced liposarcoma of the retropharyngeal space. In: Otolaryngol Head Neck Surg 134, 2006, S. 1060–1062. PMID 16730558
- Z. Orosz u. a.: Pleomorphic liposarcoma of a young woman following radiotherapy for epithelioid sarcoma. (PDF; 190 kB) In: Pathol Oncol Res 6, 2000, S. 287–291. PMID 11173662
- T. A Nickloes u. a.: Lipomas. Vom 16. März 2010
- R. D. Brasfield und T. K. Das Gupta: Liposarcoma. In: CA Cancer J Clin 20, 1970, S. 3–8. PMID 5005753
- W. Remmele u. a.: Pathologie (Kopf-Hals-Region, Weichgewebstumoren, Haut) 3. Auflage, Verlag Springer, 2008. Auszug in der Google-Buchsuche
- M. B. Binh u. a.: MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing well-differentiated and dedifferentiated liposarcoma subtypes: a comparative analysis of 559 soft tissue neoplasms with genetic data. In: Am J Surg Pathol 29, 2005, S. 1340–1347. PMID 16160477
- N. Sirvent u. a.: Detection of MDM2-CDK4 amplification by fluorescence in situ hybridization in 200 paraffin-embedded tumor samples: utility in diagnosing adipocytic lesions and comparison with immunohistochemistry and real-time PCR. In: Am J Surg Pathol 31, 2007, S. 1476–1489. PMID 17895748
- R. L. Jones u. a.: Differential sensitivity of liposarcoma subtypes to chemotherapy. In: Eur J Cancer 41, 2005, S. 2853–2860. PMID 16289617
- R. A. Schwartz u. a.: Liposarcoma: Treatment and Medication. Vom 18. April 2008
- R. A. Schwartz u. a.: Liposarcoma: Follow-up. Vom 18. April 2008
Weblinks
- Liposarcoma (englisch)
- Histopathology India: Liposarcoma (englisch)