Langerhans-Zell-Histiozytose
Die Langerhans-Zell-Histiozytose (Abkürzung: LCH; früher: Histiozytose X; englisch: histiocytosis X, langerhans-cell histiocytosis) oder Langerhanszell-Granulomatose ist eine Erkrankung aus der Gruppe der Histiozytosen.[1] Man unterscheidet drei Verlaufsformen, wobei eine exakte Zuordnung meist schwierig ist. Die Inzidenz liegt zwischen 1/200.000 und 1/2.000.000. Ein Überwiegen beim männlichen Geschlecht wird angenommen. Die Einteilung in Risikogruppen und die daraus resultierende Therapie-Indikation erfolgt heute nach dem sogenannten Lipton-Score.
| Klassifikation nach ICD-10 | |
|---|---|
| C96.0 | Multifokale und multisystemische (disseminierte) Langerhans-Zell-Histiozytose [Abt-Letterer-Siwe-Krankheit] inklusive: Histiozytose X, multisystemisch |
| C96.5 | Multifokale und unisystemische Langerhans-Zell-Histiozytose inklusive: Hand-Schüller-Christian-Krankheit; Histiozytose X, multifokal |
| C96.6 | Unifokale Langerhans-Zell-Histiozytose inklusive: Eosinophiles Granulom; Histiozytose X, unifokal; Histiozytose X o.n.A.; Langerhans-Zell-Histiozytose o.n.A. |
| ICD-10 online (WHO-Version 2019) | |
Pathologie
Bei der LCH leiten sich die Tumorzellen von Langerhans-Zellen ab. Diese stammen aus dem Knochenmark und werden den klassischen Gewebsmakrophagen und damit dem mononukleären Phagozyten-System (MPS) zugeordnet. Im Elektronenmikroskop lassen sich häufig Birbeck-Granula (tennisschlägerartige, am Stiel pentalaminäre Granula) nachweisen. Immunhistochemisch sind die Tumorzellen S-100, Vimentin und CD1a (auf der Zelloberfläche) positiv. Unter dem Elektronenmikroskop lassen sich die Birbeck-Granula als X-Körperchen erkennen. Daher stammte das „X“ in der früheren Namensgebung. Die Morphologie der Läsionen kann je nach Alter der Läsion variieren. Frühe Läsionen zeigen dabei meist viele typische und proliferierende Langerhans-Zellen. Mit dem Alter der Läsion werden die Langerhans-Zellen weniger. Selten findet man in den Läsionen gar keine Langerhans-Zellen oder Nekrosen.
Diagnostik
Die Erkrankung tritt durch Schmerzen im Kindes- und jungem Erwachsenenalter in Erscheinung. Nicht selten findet sich an oberflächlich lokalisierten Knochen (Rippe, Schädel) eine teigige Schwellung. Die LCH gilt oft als Krebs durch das unkontrollierte Zellwachstum. Auch weist sie Merkmale von Autoimmunerkrankungen auf, da LCH-Läsionen Immunzellen anziehen und typische Gewebe-Entzündungen aufweisen. LCH ist klinisch vielfältig und oft schwer zu diagnostizieren. Erkrankungen mit Hautbeteiligung bei Säuglingen können wie ein Windelausschlag aussehen, während eine Knochenbeteiligung im Röntgenbild als Sarkom verwechselt werden kann. In der aggressivsten Form kann LCH als Leukämie-ähnliche Krankheit auftreten und zu Organversagen führen.
Im Röntgenbild steht eine Osteolyse im Vordergrund, ein Befall der Lunge lässt sich vor allem in der Computertomographie durch das „Cheerio-Zeichen“ erkennen.[2][3] An den Extremitäten ist die Diaphyse bevorzugt betroffen, sonst kurze platte Knochen wie relativ häufig der Schädelknochen. Diagnostisch wegweisend ist der Nachweis eines Plattwirbels, Vertebra plana.
Im Ultraschall lässt sich die Weichteilkomponente sowie die knöcherne Destruktion gut nachweisen, charakteristisch ist an der Kalotte die gestufte unterschiedliche Destruktion im Knochen (Tabula externa).
Die Diagnose wird in der Regel durch eine Biopsie aus einem Tumor gesichert (vgl. Pathologie). LCH muss von Nicht-Langerhans-Zell-Histiozytosen (Klasse-II-Histiozytosen) abgegrenzt werden.
Formen
Man unterscheidet drei Krankheitsentitäten innerhalb einer Entwicklungshierarchie hinsichtlich Verlauf, Prognose und klinischer Präsentation, die oft nicht exakt zu trennen sind.[4]
Differenzierung in
- eosinophiles Granulom: 5.–20. Lebensjahr
- Hand-Schüller-Christian-Granulomatose: 3.–5. Lebensjahr, chronischer und manchmal tödlicher Verlauf
- Abt-Letterer-Siwe-Granulomatose: 1.–3. Lebensjahr, rascher und tödlicher Verlauf[5]
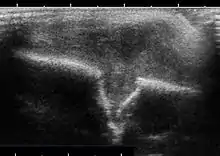
Eosinophiles Granulom
Das unifokale bzw. multifokale eosinophile Granulom ist die lokalisierte Verlaufsform der Langerhans-Zell-Histiozytose. Es ist deren häufigste Verlaufsform und macht ca. 70 % der Fälle aus.
Patienten
Meist handelt es sich bei den Patienten um Kinder oder junge Erwachsene unter 30 Jahren.
Symptome und Befallsmuster
_S-100.JPG.webp)
Hier finden sich meist in der Markhöhle der Knochen (häufig Oberschenkel, Schädelknochen und Rippen) multifokale bzw. unifokale Tumoren aus erosiv expandierenden und zum Teil mehrkernigen Langerhans-Zellen. Es findet sich ein teils belastungsabhängiger, teils auch nächtlicher lokalisierter Knochenschmerz. In der Regel findet sich ein buntes Begleit-Infiltrat aus Eosinophilen, Neutrophilen, Lymphozyten und Plasmazellen. Außerdem können sich Tumoren in der Lunge, der Haut und dem Magen manifestieren.
- Unifokale Tumoren betreffen meist die Knochen. Sie können schmerzhaft sein und pathologische Frakturen verursachen. Die Tumoren verschwinden zum Teil von selbst oder nach lokaler Exzision beziehungsweise Bestrahlung oder Chemotherapie.
- Multifokale Tumoren betreffen meist Kinder.
Verlauf
Unifokale Herde können sich spontan zurückbilden oder exzidiert bzw. bestrahlt werden. Multifokale Herde sind im Verlauf variabel und je nach betroffenem Organ unterschiedlich (vgl. Lipton-Score).
Abt-Letterer-Siwe-Syndrom
Das Abt-Letterer-Siwe-Syndrom ist die akute und disseminierte Verlaufsform der LCH. Sie ist die schwerste Form und macht etwa 10 % der Langerhans-Zell-Histiozytosen aus.
Patienten
Die Patienten sind in der Regel jünger als zwei Jahre. Selten sind Erwachsene betroffen.
Symptome und Befallsmuster
- schuppende, ekzematoide und seborrhoische Hautinfiltrate mit Betonung von Bauch, Thorax, Rücken, Hals, Knie, Ellenbogen und Kopf
- Fieber
- Lymphknoteninfiltration mit Lymphknotenschwellungen (Lymphadenopathie)
- Infiltration von Leber und Milz mit Splenomegalie bzw. Hepatosplenomegalie
- Anorexie
- Infiltration des Knochenmarks mit der Folge:
- Anämie
- Thrombozytopenie mit der Folge einer Blutungsneigung
- Granulozytopenie bzw. Neutropenie mit der Folge einer erhöhten Anfälligkeit für Infekte (z. B. Mastoiditis, Otitis media)
- Lungensymptome (Husten, Tachypnoe, durch Exsudat bedingter Pleuraerguss[6] u. a.)
Verlauf
Unbehandelt verläuft die Erkrankung rasch tödlich (bei 90 %). Unter intensiver Chemotherapie, evtl. mit Stammzelltransplantation, ist die Überlebensrate deutlich besser.
Patienten
Das Hand-Schüller-Christian-Syndrom befällt meist 2–5-jährige Kinder, Jugendliche und Erwachsene mittleren Alters. Diese Form macht etwa 15–40 % der Langerhans-Zell-Histiozytosen aus.
Symptome und Befallsmuster
Die überwiegend aus Langerhans-Zellen bestehenden Infiltrate liegen meist im Bereich der Schädelknochen und der Schädelweichteile.
- Skelettveränderungen v. a. bei flachen Knochen (Schädels(Landkartenschädel), Rippen, Schulterblatt (Scapula), Becken)
- Exophthalmus bei Befall der Hypophyse und des Sehnervs
- Strabismus und Sehverlust bei Befall des Nervus opticus und der Augenmuskeln
- Zahnverlust bei Infiltration des Zahnfleisches (Gingiva) und des Unterkiefers (Mandibula)
- Chronische Otitis media und externa, Mastoiditis bei Befall des Innenohres
- Bei Hypophyseninfiltration:
- Diabetes insipidus bei Befall der Hypophyse und des Hypothalamus (bei 5–50 % der Patienten)
- Wachstumsstörungen bei Befall der Hypophyse (bei ca. 40 % der Patienten)
- Hyperprolaktinämie
- Hypogonadismus
Bei etwa 30 % der betroffenen Menschen kommt es zum systemischen Befall mit Einbeziehung von Leber, Milz, Lungen, Haut und Lymphknoten. Die klassische Hand-Schüller-Christian-Trias mit Knochenläsionen, Exophthalmus und Diabetes insipidus tritt eher selten auf.
Verlauf
Bei systemischem Befall multipler Organe besteht eine schlechte Prognose und die Notwendigkeit einer aggressiven Chemotherapie und evtl. einer Stammzelltransplantation. Ansonsten kann sich die Erkrankung von selbst zurückbilden.
Literatur
- S1-Leitlinie Langerhanzell-Histiozytose (LCH) im Kindes- und Jugendalter der Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH). In: AWMF online (Stand Juni 2012)
- Jürgen Freyschmidt, H. Ostertag, G. Jundt: Knochentumoren. Springer, 2003, ISBN 3-540-40364-7.
Weblinks
Einzelnachweise
- Joachim Fichter u. a.: Langerhans-Zellhistiozytose des Erwachsenen – eine interdisziplinäre Herausforderung. In: Deutsches Ärzteblatt. Nr. 104, 2007, S. 2347 (aerzteblatt.de).
- D. Aktuerk, M. Lutz, D. Rosewarne, H. Luckraz: Cheerios in the lung: a rare but characteristic radiographic sign. In: QJM: An International Journal of Medicine. Band 108, September 2015, S. 743–744, doi:10.1093/qjmed/hcv044, PMID 25660601.
- S. H. Chou, G. Kicska, J. P. Kanne, S. Pipavath: Cheerio sign. In: Journal of Thoracic Imaging. Band 28, Januar 2013, S. W4, doi:10.1097/RTI.0b013e31827944d2, PMID 23254591.
- F Halbritter et al.: Epigenomics and Single-cell Sequencing Define a Developmental Hierarchy in Langerhans Cell Histiocytosis. In: Cancer Discovery. Juli 2019, doi:10.1158/2159-8290.CD-19-0138.
- B. Hansen, P. Schwarz: Histiocytosis X. Review of the literature and a case report. In: Ugeskr Laeger. 151(1), 2 Jan 1989, S. 5–7
- Berthold Jany, Tobias Welte: Pleuraerguss des Erwachsenen – Ursachen, Diagnostik und Therapie. In: Deutsches Ärzteblatt. Band 116, Nr. 21, 2019, S. 377–385, hier: S. 379 f.