Morbus Behçet
Der Morbus Behçet (türkische Aussprache: ˈbɛhtʃæt ‚beHtschett‘ (hörbares „h“), auch Behçet-Krankheit, Morbus Adamantiades-Behçet (ABD) oder maligne Aphthose genannt) ist eine systemische, autoimmune Entzündung der Blutgefäße (Vaskulitis). Sie gehört damit zum Rheumatischen Formenkreis. Es sind vor allem kleine Venen und Kapillaren betroffen, grundsätzlich können aber alle Blutgefäße jeder Größe befallen werden. Da die Entzündungen in jedem Organsystem auftreten können, ist beim Morbus Behçet ein breites Spektrum an Symptomen möglich. Als typisch gelten die Kombination von Aphthen im Mund und im Intimbereich sowie Entzündungen von Strukturen des Auges. Das Hauptziel der Therapie ist die Verhinderung von Folgeschäden, die insbesondere bei Beteiligung der Augen, des Gehirns und großer Gefäße schwerwiegend sein können. Dazu wird mit Immunsuppressiva die Aktivität des Immunsystems gedämpft.
| Klassifikation nach ICD-10 | |
|---|---|
| M35.2 | Behçet-Krankheit |
| ICD-10 online (WHO-Version 2019) | |

Die Ursache der Erkrankung ist nicht bekannt. Nach dem heutigen Erkenntnisstand führt bei bestimmten genetischen Voraussetzungen (insbesondere das Vorliegen des HLA-Typs B51) der Kontakt mit einem äußeren Faktor (zum Beispiel der Kontakt mit einem Virus oder Bakterium) zu einer Überreaktion des Immunsystems und zu einer unkontrollierten Entzündung. Die Krankheit tritt vor allem in den Ländern der historischen Seidenstraße auf, ist durch Migration aber auch in Europa und Nordamerika häufiger anzutreffen. Das Geschlechterverhältnis der Erkrankten ist regional unterschiedlich. Der Erkrankungsgipfel liegt weltweit zwischen 20 und 40 Jahren. Die Krankheit verläuft wellenförmig mit Phasen stärkerer und schwächerer Aktivität, klingt aber im Allgemeinen mit zunehmender Erkrankungsdauer ab.
Benannt ist die Erkrankung nach dem türkischen Hautarzt Hulusi Behçet (1889–1948) und dem griechischen Augenarzt Benediktos Adamantiades (1875–1962), die die Symptome der Erkrankung unabhängig voneinander in den 1930er-Jahren beschrieben.
Verbreitung
Der Morbus Behçet ist vor allem entlang der Seidenstraße im Nahen und Mittleren Osten sowie in Ostasien verbreitet. Durch Migration tritt die Krankheit auch in Europa und Nordamerika häufiger auf. Für die einzelnen Länder gibt es unterschiedliche Angaben zur Häufigkeit. Das Land mit der höchsten Prävalenz ist die Türkei mit 20 bis an manchen Orten 420 Erkrankten auf 100.000 Einwohner. In Iran liegt die Prävalenz bei 17–68 pro 100.000, in Japan bei 7–14 pro 100.000. Für Deutschland finden sich Angaben zwischen 0,6 und 1,5 auf 100.000 Einwohner.[1] Bei Studien an Migranten ergaben sich folgende Befunde: Armenier, die in Istanbul leben, haben ein deutlich niedrigeres Erkrankungsrisiko als die Allgemeinbevölkerung der Türkei. In Deutschland lebende Türken haben ein deutlich höheres Erkrankungsrisiko als die deutsche Allgemeinbevölkerung, die Prävalenz unter den in Deutschland lebenden Türken ist aber geringer als die Prävalenz in der Türkei.[2]
Das Geschlechterverhältnis unterscheidet sich regional. In einigen Ländern sind Frauen häufiger betroffen, besonders deutlich ist der Unterschied in Schottland, den Vereinigten Staaten und in Spanien. Auch in Israel, Schweden, Korea und Japan überwiegen die Frauen. Zu den Ländern, in denen Männer häufiger betroffen sind, gehören unter anderem die Türkei (allerdings ist hier der Überhang nur leicht), Deutschland, Italien, Frankreich und Iran. Sehr stark ist der Unterschied im Irak, in Saudi-Arabien, Russland und Kuwait.[3]
Das mittlere Erkrankungsalter liegt in den meisten Ländern in der dritten Lebensdekade. Die Spanne liegt zwischen 20 Jahren in Irland und 40 Jahren in Brasilien. In der Türkei und in Deutschland liegt das mittlere Erkrankungsalter bei 25 bis 26 Jahren. In den USA und den asiatischen Ländern erkranken die Patienten im Alter von über 30 Jahren.[3]
Ursachen
Als Ursachen der Krankheitsentstehung (Ätiologie) können genetische Voraussetzungen und Umweltfaktoren angenommen werden, was sich insbesondere aus den Erkenntnissen der Studien an Personen mit Migrationsgeschichte ableitet. Eine Möglichkeit, Genvarianten zu identifizieren, die mit der Krankheit in Verbindung stehen (assoziiert sind), sind Genomweite Assoziationsstudien.
Human Leukocyte Antigen (HLA) ist die Bezeichnung für Proteine, die auf der Oberfläche von Zellen sitzen und dem Immunsystem Teile anderer in der Zelle produzierter Proteine präsentieren. Entsprechen die präsentierten Teile nicht dem üblichen Bauplan, erkennt das Immunsystem dies als „fremd“ und greift die Zelle an. Von den HLA-Proteinen existieren viele Varianten, von denen manche mit der Entstehung von Autoimmunerkrankungen in Verbindung stehen. Der stärkste Zusammenhang besteht beim Morbus Behçet zum HLA-B51. Dieser HLA-Typ findet sich bei bis zu 60 % der Erkrankten.[4] Daneben wurden weitere HLA-Typen und Varianten von Genen, die für die Funktion des Immunsystems wichtig sind, identifiziert. Unter ihnen ist auch das Gen für Interleukin-10, ein anti-entzündlicher Botenstoff, und der Rezeptor für Interleukin 23.[5]
Bislang konnte kein infektiöser Auslöser nachgewiesen werden. Diskutiert wurden Streptococcus sanguis, Herpes-simplex-Viren, das Epstein-Barr-Virus und Cytomegalieviren. Es konnte nachgewiesen werden, dass das Immunsystem von Behçet-Patienten stärker auf Antigene von Streptococcus sanguis reagiert, aber eine direkte Verbindung zur Krankheit konnte nicht gezeigt werden.[5]
Pathologie und Krankheitsentstehung
In der mikroskopischen Untersuchung erkrankter Blutgefäße zeigt sich das Bild einer sogenannten leukozytoklastischen Vaskulitis. Das bedeutet, dass Leukozyten (weiße Blutkörperchen, die Immunzellen) in das Gewebe einwandern und dieses zerstören.[6] Die Leukozyten-Typen, die dabei gefunden werden, sind je nach Gefäßart unterschiedlich. Es finden sich häufig diffuse Nekrosen und Transsudate in den entzündeten Gefäßwänden. Im umliegenden Gewebe zeigen sich Ödeme und Ansammlungen von Makrophagen.[7]
Es gibt noch kein schlüssiges Modell der Krankheitsentstehung. Es ist gesichert, dass beide Anteile des Immunsystems an den Krankheitsprozessen beteiligt sind, denn es finden sich Anzeichen der angeborenen und der erworbenen Immunantwort. Wahrscheinlich ist die Krankheit durch T-Helferzellen vermittelt, die dem adaptiven Immunsystem zuzuordnen sind. Diesen Immunzellen werden von anderen Zellen Antigene präsentiert. Ihre Aufgabe ist es, zu entscheiden, ob es sich dabei um „fremde“ Antigene handelt und ob eine Immunantwort eingeleitet wird. Bei Behçet-Patienten mit ihren entsprechenden genetischen Voraussetzungen reagieren die TH1-Lymphozyten (ein Typ der T-Helferzellen, der das angeborene Immunsystem aktiviert) zu stark auf eine Aktivierung. Dabei setzen sie eine sich selbst aufrechterhaltende Signalkaskade in Gang, die zu einer unkontrollierten Antwort des angeborenen Immunsystems führt.[7][8]
Die Einordnung des Morbus Behçet ist nicht einfach: Als Autoimmunerkrankung (wie zum Beispiel der Morbus Basedow) gilt er nicht, da bislang kein gegen körpereigene Strukturen gerichteter Autoantikörper bekannt ist. Den autoinflammatorischen Krankheiten (wie das Familiäre Mittelmeerfieber) ist er auch nicht zuzuordnen. Hier wäre das angeborene Immunsystem der Verursacher, aber die typischen genetischen Voraussetzungen und Symptome wie wiederkehrende Fieberschübe fehlen.[8]
Symptome
| Manifestationsort | Häufigkeit in Prozent |
|---|---|
| Orale Aphthen | 47–86 |
| Genitale Aphthen | 57–93 |
| Auge | 30–70 |
| Haut | 38–99 |
| Gelenke | 45–60 |
| Herz-Kreislauf-System | 7–49 |
| Nervensystem | 5–10 |
| Magen-Darm-Trakt | 3–26 |
Da es sich beim Morbus Behçet um eine Systemerkrankung handelt, die potentiell jedes Organsystem betreffen kann, ist das klinische Bild sehr variabel. Als typisch für die Erkrankung gilt die Trias (das Auftreten von drei Symptomen) aus Aphthen im Mund und im Intimbereich sowie einer Uveitis.[9]
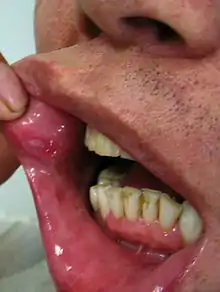
Orale und genitale Geschwüre
Das häufigste Frühsymptom des Morbus Behçet sind wiederkehrende Aphthen im Mund. Sie werden bei allen Patienten im Krankheitsverlauf beobachtet. Sie treten einzeln oder gehäuft auf und betreffen meistens die Wangenschleimhaut, das Zahnfleisch, die Lippen und die Zunge. Gaumen und Rachen sind seltener betroffen. Kleinere Aphthen (<1 cm Durchmesser) heilen in der Regel innerhalb von 4 bis 14 Tagen ab, größere brauchen länger.[9]
Geschwüre im Intimbereich sind ebenfalls häufig. Sie sind in der Regel tiefer und schmerzhafter als die oralen Aphten und heilen mit Narbenbildung ab. Bei Frauen treten sie an der Vulva, in der Vagina und am Gebärmutterhals auf. Bei Männern vor allem am Hodensack, der Penis ist nur selten betroffen. Bei beiden Geschlechtern können die Geschwüre am Damm, rund um den Anus oder in der Leistenregion auftreten.[9]
Augenbeteiligung
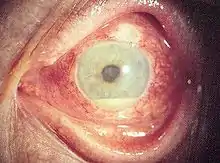
Bei rund 10 % der Patienten ist ein Auge das erste betroffene Organ. Das Spektrum der Manifestationen ist weit: Typischerweise treten Uveitiden auf, deren äußeres Merkmal oft eine Eiteransammlung in der vorderen Augenkammer (Hypopyon) ist. Außerdem können unter anderem Entzündungen der Netzhaut, der Skleren, der Hornhaut und des Sehnerven auftreten. Als Beschwerden können beispielsweise Schmerzen der Augen, Einschränkungen der Sehfähigkeit, Lichtscheu und verstärkter Tränenfluss bestehen. Generell ist eine Augenbeteiligung eher bei Männern zu finden und mit einem schwereren Krankheitsverlauf assoziiert.[9][10]
Hautbeteiligung
Der Befall der Haut kann sich äußern in papelartigen Veränderungen, Erythema-nodosum-artigen Läsionen, akneiforme Ausschlägen, Pyoderma gangraenosum oder seltener in Erythema-multiforme-artigen Ausschlägen. Unter Pathergie versteht man eine Hyperreaktivität der Haut. Diese reagiert auf kleine Verletzungen wie Nadelstiche zeitverzögert mit einem papulopustulösen Ausschlag. Dieses Phänomen kann zur Diagnostik herangezogen werden, tritt aber nicht nur beim Morbus Behçet auf.[11]
Gelenkbeteiligung
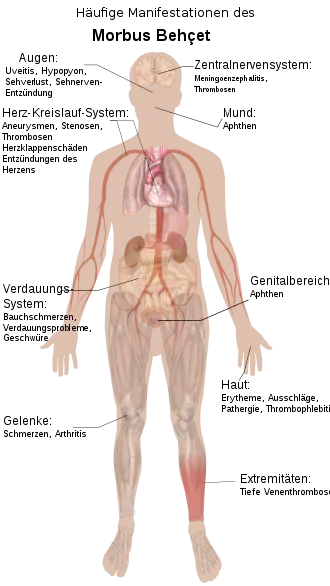
45 bis 60 % der Patienten haben Manifestationen an den Gelenken. Dabei treten Gelenkschmerzen auf oder Entzündungen eines oder mehrerer Gelenke. Üblicherweise sind Knie, Knöchel, Ellbogen und Handgelenke betroffen. Die Entzündungen sind nicht gelenkzerstörend.[9] Im Krankheitsverlauf kann sich vor allem bei Frauen ein Fibromyalgiesyndrom entwickeln. In manchen Studien wurden auch Entzündungen des Kreuzbein-Darmbein-Gelenks und eine ankylosierende Spondylitis bei Behçet-Patienten gefunden.[12] In Fachbüchern werden diese Symptome aber nicht zu den Behçet-Manifestationen gezählt.[13]
Beteiligung des Herz-Kreislauf-Systems
Da der Morbus Behçet eine Entzündung der Gefäße ist, entstehen auch an diesen direkte Schäden. Venen sind die am häufigsten betroffenen Blutgefäße. Ihre Entzündung (Phlebitis) verursacht häufig die Bildung von Blutgerinnseln. Je nach Lage der entzündeten Venen zeigen sich Thrombophlebitiden (bei oberflächlichen Venen) oder tiefe Venenthrombosen (in den tief liegenden Venen der Arme und Beine). Selten treten Thrombosen in den Hohlvenen, im Durasinus oder in den Lebervenen auf.
Eine Erkrankung der Arterien ist seltener und eher bei Männern anzutreffen. Sie verursacht Aussackungen der Arterienwände (Aneurysmen), Gefäßverengungen und ebenfalls Thrombosen.[14] Eingerissene Aneurysmen verursachen Blutungen, die je nach Ort schwere Folgen haben können (beispielsweise bei Aussackungen an den Lungenarterien, die allerdings sehr selten sind).[9]
Die Erkrankung kann auch das Herz direkt betreffen, dies ist allerdings selten. Manifestationen umfassen neben der Schädigung der Herzkranzgefäße auch Entzündungen des Herzbeutels (Perikarditis), des Herzmuskels (Myokarditis) und der Herzinnenhaut (Endokarditis) sowie Schädigungen der Herzklappen.[14]
Nervensystem
Bei rund 10 % der Patienten entwickelt sich einige Jahre nach Beginn der Erkrankung eine Erkrankung des Nervensystems, die auch Neuro-Behçet genannt wird. Sie betrifft in der Regel das Zentralnervensystem und nur selten das periphere Nervensystem. Üblicherweise wird der Neuro-Behçet in zwei Formen eingeteilt: die parenchymale und die nicht-parenchymale Form. Die parenchymale Form ist keine Entzündung der Blutgefäße, sondern des Hirngewebes um die Blutgefäße herum (eine sogenannte Perivaskulitis). Die nicht-parenchymale Form ist dagegen die Entzündung der Blutgefäße des Gehirns (also die dem Morbus Behçet zugrundeliegende Vaskulitis). Der überwiegende Teil der Patienten mit Neuro-Behçet leidet an der parenchymalen Form. Sie präsentiert sich als Meningoenzephalitis (also eine Entzündung von Gehirn und Hirnhäuten) mit Migräne-ähnlichen Kopfschmerzen. Sie trifft oft den Hirnstamm und die Basalganglien. Möglich sind motorische, sensible und kognitive Einschränkungen (wie Konzentrationsstörungen). Die Folgen der nicht-parenchymalen Form umfassen Thrombosen in den Durasinus und Hirnvenen, Aneurysmen der Arterien und erhöhten Hirndruck. Ischämische Schlaganfälle sind selten. Typischerweise entwickelt sich ein Neuro-Behçet bei Männern und geht mit einer Phase erhöhter Krankheitsaktivität einher.[15]
Gastrointestinale Beteiligung
Entzündungen des Magen-Darm-Trakts sind vor allem bei Patienten in Japan zu beobachten, in den anderen Regionen der Welt sind sie eine seltenere Manifestation des Morbus Behçet. Geschwüre können im gesamten Magen-Darm-Trakt auftreten. Am häufigsten entstehen sie im Endabschnitt des Dünndarms (Ileum, Ileitis) und im Blinddarm (Coecum). Anzeichen für eine Beteiligung des Magen-Darm-Trakts sind Bauchschmerzen, Appetitverlust, Erbrechen, Durchfall und Teerstuhl. Selten brechen die Geschwüre durch (Perforation).[9]
Andere Symptome
Bei Männern können auch Nebenhodenentzündungen Zeichen der Erkrankung sein. Selten sind die Nieren in Form einer Glomerulonephritis oder einer Amyloidose betroffen. Bei Beteiligung der Harnblase treten Blasenentleerungsstörungen auf. Bei einigen Patienten wurden wiederkehrende Knorpelentzündungen in Verbindung mit anderen Behçet-Symptomen beschrieben (MAGIC-Syndrom: Entzündungen des Knorpels der Nase und der Ohrmuscheln mit oralen und genitalen Aphthen).[16]
Diagnose
Die Diagnosestellung wird durch das breite Spektrum möglicher Symptome, die variable Präsentation der Krankheit und das Fehlen von Biomarkern erschwert. Die Diagnose muss daher nach klinischen Symptomen bei gleichzeitigem Ausschluss anderer möglicher Ursachen erfolgen. Es sind drei Klassifikationssysteme in Gebrauch. Das älteste sind die Kriterien der International Study Group von 1990. In diesem System müssen das Hauptsymptom der oralen Aphthen und zwei Nebensymptome (genitale Aphten, Augen- oder Hautsymptome oder eine Pathergie-Reaktion nach Injektion von 0,5 ml Kochsalzlösung in die Haut) erfüllt sein. 2006 wurden die International Criteria for Behçet’s Disease (ICBD) publiziert, die 2014 überarbeitet wurden. Darin ist jedem Symptom ein Punktwert zugeordnet. Die Punktwerte der aufgetretenen Symptome werden addiert, bei Erreichen eines bestimmten Punktwertes kann das Vorliegen des Morbus Behçet angenommen werden.[11][17] Pathognomonisch ist das kutane Pathergiephänomen, bei dem auftretende Pusteln im Schub an der Injektionsstelle einer 0,9-prozentigen NaCl-Lösung auftreten.[18] Der Test wird sowohl kutan (an der Innenseite eines Unterarms) als auch oral (an der Innenseite der Unterlippe) durchgeführt.
Differentialdiagnose
Unter Differenzialdiagnosen werden Krankheiten verstanden, die ein ähnliches Beschwerdebild zeigen und daher im Diagnoseprozess ausgeschlossen werden müssen. Bei Morbus Behçet sind dies vor allem andere rheumatische Systemerkrankungen wie Vaskulitiden, Systemischer Lupus erythematodes und Sarkoidose. Für die einzelnen Manifestationsformen kommen unterschiedliche Krankheiten differenzialdiagnostisch in Frage:[19]
- Die oralen Aphthen des Morbus Behçet müssen von einer schweren Verlaufsform der benignen Aphthose, mit zwar ebenso schmerzhaften, aber harmlosen Aphthen ohne systemische Erkrankung, abgegrenzt werden, der Aphthosis vom Typ Mikulicz, vom Typ Sutton und vom Typ Cooke.[20] Außerdem können Infektionen mit Herpes simplex oralis und Aphthen aufgrund von Mangelzuständen (wie sie bei Zöliakie oder chronischen Darmerkrankungen auftreten können) in Betracht kommen.
- Bei genitalen Aphthen können sexuell übertragbare Erkrankungen ursächlich sein.
- Die Augenbeteiligung in Form der Uveitis kann zahlreiche andere Ursachen haben, unter ihnen Infektionen mit Viren, Bakterien und Pilzen, oder Symptom einer anderen systemischen Erkrankung sein (siehe Uveitis: Sekundäre Formen)
- Bei Befall des Verdauungssystems muss vor allem Morbus Crohn ausgeschlossen werden.
- Der Neuro-Behçet muss von der Multiplen Sklerose abgegrenzt werden.
- Manifestationen am Skelettsystem müssen von anderen Gelenkerkrankungen unterschieden werden: Ankylosierende Spondylitis, Rheumatoide Arthritis ohne Erhöhung der typischen Laborparameter (seronegative Arthritis) und die Psoriasisarthritis (ebenfalls eine seronegative Arthritis).
- Das Hughes-Stovin-Syndrom ist eine Erkrankung, die mit Thrombosen und Aneurysmen der Lungenarterien einhergeht. Es ist allerdings unklar, ob es sich um eine eigenständige Krankheit handelt oder eine Ausprägung des Morbus Behçet.
In westlichen Ländern sind der Morbus Crohn und die seronegativen Arthritiden die Haupt-Differentialdiagnosen des Morbus Behçet. Insbesondere wenn nur eine Manifestationsform vorliegt, kann die Unterscheidung unmöglich sein.[19]
Verlauf und Prognose
Die Krankheit hat einen wellenförmigen Verlauf mit Phasen zu- und abnehmender Krankheitsaktivität. Grundsätzlich klingt sie mit zunehmender Erkrankungsdauer ab.[2]
Viele Behçet-Manifestationen heilen ohne bleibende Schäden ab.[21] Männer und in jungen Jahren Erkrankte haben schwerere Verläufe und eine höhere Sterblichkeit. Die höchste Sterblichkeitsrate haben demnach junge Männer, die zwischen 14 und 24 Jahren diagnostiziert wurden. Mit der Zeit sinkt aber auch ihre Sterblichkeit. Haupttodesursachen sind der Neuro-Behçet, die gastro-intestinale Beteiligung sowie Aneurysmen der Lungenarterien, die bei jedem fünften davon Betroffenen innerhalb von fünf Jahren zum Tod führen (5-Jahres-Mortalität 20 %). Auch Verschlüsse der Lebervenen, das Budd-Chiari-Syndrom, haben eine schlechte Prognose.[19] Die Augenbeteiligung geht mit einem hohen Risiko für Erblindung einher – rund 25 bis 50 % der betroffenen Patienten verlieren ihr Sehvermögen. Insgesamt aber hat sich die Prognose, vor allem das Erblindungsrisiko, in den letzten Jahren deutlich verbessert.[22]
Auch in weniger schweren Fällen hat die Erkrankung erheblichen Einfluss auf das körperliche und seelische Befinden der Betroffenen. Die Erkrankung beeinträchtigt zwar nicht direkt die Fruchtbarkeit, diese steht aber unter dem Einfluss der Auswirkungen genitaler Geschwüre und der psychischen Belastung durch die Krankheit. Es gibt keine Hinweise, dass sich die Erkrankung negativ auf die Schwangerschaft auswirkt.[23]
Therapie
Da die Erkrankung häufig in ihrer Anfangsphase am schwersten ist und mit der Zeit ausklingt, ist die Therapie tendenziell nach der Erstdiagnose am intensivsten, während im späteren Krankheitsverlauf für viele Patienten keine Therapie mehr notwendig ist. Es gibt keine Standardtherapie: welche Mittel gewählt werden, ist von der Schwere der Symptome, den betroffenen Organsystemen und auch den örtlichen Gegebenheiten in den verschiedenen Ländern abhängig.[24] Gute Therapiestudien fehlen häufig, sodass viele Empfehlungen auf Expertenmeinungen beruhen. Grundbaustein der medikamentösen Therapie ist die Beeinflussung des Immunsystems. Generell gilt, dass Entzündungen der Netzhaut, arterielle Aneurysmen, arterielle Vaskulitis und der Neuro-Behçet wegen ihrer potentiell schweren Folgen aggressiver therapiert werden. Zur Unterdrückung des Immunsystems (Immunsuppression) werden unter anderem Glucocorticoide wie Prednisolon und andere Immunsuppressiva wie Azathioprin, Mycophenolat-Mofetil, Ciclosporin und Methotrexat, TNF-Blocker wie Infliximab und andere monoklonale Antikörper wie Rituximab und Alemtuzumab genutzt.[25] Bei Aphthen und Gelenkentzündungen wird häufig Colchicin genutzt. Eine neue Therapieoption ist Apremilast, das als eines von wenigen Medikamenten in einer Phase-II-Studie erprobt[26] wurde und Wirksamkeit zeigte. Bei Beteiligung des Magen-Darm-Traktes wird häufig in Analogie zum Morbus Crohn Mesalazin eingesetzt.[24] Neben der von der Krankheitsausprägung abhängigen immunsuppressiven Therapie können weitere Maßnahmen wie Schmerztherapie oder die lokale Behandlung von Hautsymptomen und der Aphthen sinnvoll sein.[27] Zur Anwendung kommen auch Cyclophosphamid, Interferone und Dapson.[18] Bei schwerwiegenden Komplikationen, also rasch wachsende Aneurysmen und die Folgen von Fisteln und Perforationen im Magen-Darm-Trakt, können operative Eingriffe notwendig sein. Diese sollen mit starker Immunsuppression begleitet werden.[28]
Historische Aspekte
Die Krankheit ist nach dem türkischen Hautarzt Hulusi Behçet benannt, der 1937 die drei Symptome der oralen und genitalen Aphthen mit einer Regenbogenhautentzündung mit Hypopyon als Syndrom veröffentlichte und damit als eigenständiges Krankheitsbild auffasste.[29] Allerdings beschrieb der griechische Augenarzt Benediktos Adamantiades schon 1930 einen Fall von Iritis mit rezidivierendem Hypopyon und den typischen oralen und genitalen Aphthen. Er schlug vor, die beobachteten Symptome als Syndrom anzusehen.[30] In den 1950er Jahren fügte er dem Symptomkomplex weitere Symptome wie Thrombophlebitiden hinzu.[31] Ihm zu Ehren und wegen dieser Arbeiten wurde später die Krankheit als Morbus Adamantiades-Behçet in das deutsche und in das griechische Register sowie auch in das National Registry for Rare Disorders (NORD) in den USA eingetragen (englisch Adamantiades-Behçet Disease (ABD)).[32]
Abgesehen von diesen Beschreibungen finden sich in der Medizingeschichte immer wieder Beschreibungen von Krankheitsfällen, die dem Morbus Behçet zugeordnet werden können. Die älteste stammt vermutlich aus der Antike von Hippokrates von Kos.[33]
Weblinks
- S2k-Leitlinie (Langversion), Diagnostik und Therapieoptionen von Aphthen und aphthoiden Läsionen der Mund- und Rachenschleimhaut, Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften (AWMF), Stand: November 2016. Abgerufen am 11. November 2017.
- www.behcet.de
- Website der Universität Tübingen zum Morbus Behçet
- Behçet’s syndrome bei whonamedit.com (englisch)
- Fallbericht eines lange unerkannten M. Behçet: Merinopoulos D, Saada J, et al.: Pattern recognition is a sequential process—accurate diagnosis and treatment 20 years after presentation. Oxford Medical Case Reports 2019 (7), Juli 2019, DOI:10.1093/omcr/omz058, mit deutschsprachiger Aufbereitung des Falls bei SPIEGEL online
Literatur
- Ina Kötter: Morbus Behçet. In: H.-H. Peter, W. J. Pichler, U. Müller-Ladner (Hrsg.): Klinische Immunologie. Urban & Fischer, München 2012, S. 399 ff.
- Jagdish R. Nair, Robert J. Moots: Behçet’s Disease. In: Clinical Medicine. 17, Nr. 1, 2017, ISSN 1470-2118, S. 71–77 (PDF-Datei; 126 kB).
- Adnan Al-Araji, Desmond P. Kidd: Neuro-Behçet’s Disease: epidemiology, clinical characteristics, and management. In: Lancet Neurology. Nr. 8, 2009, S. 192–204. (PDF, englisch).
- Gulen Hatemi et al.: 2018 update of the EULAR recommendations for the management of Behçet’s syndrome. In: Annals of the Rheumatic Diseases. Nr. 77, 2018, S. 808–818. (online, PDF)
Einzelnachweise
- Marc C. Hochberg, Alan J. Silman, Josef S. Smolen, Michael E. Weinblatt, Michael H. Weisman: Rheumatology. 6. Auflage. Band 2. Mosby/Elsevier, Philadelphia 2015, ISBN 978-0-323-09138-1, S. 1276.
- Marc C. Hochberg, Alan J. Silman, Josef S. Smolen, Michael E. Weinblatt, Michael H. Weisman: Rheumatology. 6. Auflage. Band 2. Mosby/Elsevier, Philadelphia 2015, ISBN 978-0-323-09138-1, S. 1328.
- Fereydoun Davatchi: Behcet’s disease. In: International Journal of Rheumatic Diseases. 17, 2014, S. 355, doi:10.1111/1756-185X.12378.
- Jagdish R. Nair, Robert J. Moots: Behçet’s Disease. In: Clinical Medicine. 17, Nr. 1, 2017, ISSN 1470-2118, S. 71 (PDF-Datei; 126 kB).
- Marc C. Hochberg, Alan J. Silman, Josef S. Smolen, Michael E. Weinblatt, Michael H. Weisman: Rheumatology. 6. Auflage. Band 2. Mosby/Elsevier, Philadelphia 2015, ISBN 978-0-323-09138-1, S. 1328.
- Universitätsklinikum Tübingen – Morbus Behcet. Abgerufen am 10. November 2017.
- Mohamad J. Zeidan, David Saadoun, Marlene Garrido, David Klatzmann, Adrien Six: Behçet’s disease physiopathology: a contemporary review. In: Auto-Immunity Highlights. Band 7, Nr. 1, 12. Februar 2016, ISSN 2038-0305, doi:10.1007/s13317-016-0074-1, PMID 26868128, PMC 4751097 (freier Volltext).
- Ina Kötter: Morbus Behçet. In: H.-H. Peter, W. J. Pichler, U. Müller-Ladner (Hrsg.): Klinische Immunologie. Urban & Fischer, München 2012, S. 401.
- Zeidan, Saadoun, Garrido, Klatzmann, Six, Cacoub: Behçet’s disease physiopathology: a contemporary review. In: Autoimmunity Highlights. 7, Nr. 4 2016. PMC 4751097 (freier Volltext), zuletzt abgerufen am 21. September 2017.
- Sakane, Takedo, Suzuki, Inaba: Behçet’s Disease. In: The New England Journal of Medicine 341, 1999, S. 1284–1291. (online), nicht frei zugänglich, zuletzt abgerufen am 21. September 2017.
- Jagdish R. Nair, Robert J. Moots: Behçet’s Disease. In: Clinical Medicine. 17, Nr. 1, 2017, ISSN 1470-2118, S. 72 (PDF-Datei; 126 kB).
- M. A. Ait Badi, M. Zyani, S. Kaddouri, R. Niamane, A. Hda: Les manifestations articulaires de la maladie de Behçet. À propos de 79 cas. In: La Revue de Médecine Interne. Band 29, Nr. 4, S. 277–282, doi:10.1016/j.revmed.2007.09.031.
- Marc C. Hochberg, Alan J. Silman, Josef S. Smolen, Michael E. Weinblatt, Michael H. Weisman: Rheumatology. 6. Auflage. Band 2. Mosby/Elsevier, Philadelphia 2015, ISBN 978-0-323-09138-1, S. 1330.
- Jagdish R. Nair, Robert J. Moots: Behçet’s Disease. In: Clinical Medicine. 17, Nr. 1, 2017, ISSN 1470-2118, S. 73 (PDF-Datei; 126 kB).
- Adnan Al-Araji, Desmond P Kidd: Neuro-Behçet’s disease: epidemiology, clinical characteristics, and management. In: The Lancet Neurology. Band 8, Nr. 2, 2009, S. 192–204, doi:10.1016/s1474-4422(09)70015-8.
- Marc C. Hochberg, Alan J. Silman, Josef S. Smolen, Michael E. Weinblatt, Michael H. Weisman: Rheumatology. 6. Auflage. Band 2. Mosby/Elsevier, Philadelphia 2015, ISBN 978-0-323-09138-1, S. 1330 f.
- F. Davatchi, S. Assaad-Khalil u. a.: The International Criteria for Behçet's Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. In: Journal of the European Academy of Dermatology and Venereology. 28, 2014, S. 338, doi:10.1111/jdv.12107.
- Rudolf Ott, Wolfgang Krug, Hans-Peter Vollmer: Klinik- und Praxisführer Zahnmedizin. Georg Thieme Verlag, 2002, ISBN 3-13-158021-6, S. 294 (google.com).
- Marc C. Hochberg, Alan J. Silman, Josef S. Smolen, Michael E. Weinblatt, Michael H. Weisman: Rheumatology. 6. Auflage. Band 2, Mosby/Elsevier, Philadelphia 2015, ISBN 978-0-323-09138-1, S. 1331.
- Aphthen und aphthoide Läsionen. In: Deutsche Zahnärztliche Zeitschrift. 68 (5), 2013, S. 264–268. Abgerufen am 14. November 2017.
- Fereydoun Davatchi: Behcet’s disease. In: International Journal of Rheumatic Diseases. 17, 2014, S. 355, doi:10.1111/1756-185x.12378.
- Ina Kötter: Morbus Behçet. In: H.-H. Peter, W. J. Pichler, U. Müller-Ladner (Hrsg.): Klinische Immunologie. Urban & Fischer, München 2012, S. 402.
- Jagdish R. Nair, Robert J. Moots: Behçet’s Disease. In: Clinical Medicine. 17, Nr. 1, 2017, ISSN 1470-2118, S. 76. (PDF-Datei; 126 kB)
- Yusuf Yazici, Gulen Hatemi, Bahram Bodaghi, Jae Hee Cheon, Noburu Suzuki: Behçet syndrome. In: Nature Reviews Disease Primers. Band 7, Nr. 1, 16. September 2021, ISSN 2056-676X, S. 1–14, doi:10.1038/s41572-021-00301-1 (nature.com [abgerufen am 17. November 2021]).
- Jagdish R. Nair, Robert J. Moots: Behçet’s Disease. In: Clinical Medicine. 17, Nr. 1, 2017, ISSN 1470-2118, S. 73–76 (PDF-Datei; 126 kB).
- Gulen Hatemi, Melike Melikoglu, Recep Tunc, Cengiz Korkmaz, Banu Turgut Ozturk: Apremilast for Behçet’s Syndrome — A Phase 2, Placebo-Controlled Study. In: New England Journal of Medicine. Band 372, Nr. 16, 15. April 2015, S. 1510–1518, doi:10.1056/nejmoa1408684 (nejm.org [abgerufen am 28. September 2017]).
- Jagdish R. Nair, Robert J. Moots: Behçet’s Disease. In: Clinical Medicine. 17, Nr. 1, 2017, ISSN 1470-2118, S. 74 (PDF-Datei; 126 kB).
- Jagdish R. Nair, Robert J. Moots: Behçet’s Disease. In: Clinical Medicine. 17, Nr. 1, 2017, ISSN 1470-2118, S. 75 (PDF-Datei; 126 kB).
- Türkan Saylan: Life Story of Dr. Hulusi Behçet. In: Yonsei Med J. Band 38, 1997, S. 327–332.
- C. C. Zouboulis: Benediktos Adamantiades and his forgotten contributions to medicine. In: European journal of dermatology : EJD. Band 12, Nummer 5, Sep–Okt 2002, S. 471–474. PMID 12370138.
- Ina Kötter: Morbus Behçet. In: H.-H. Peter, W. J. Pichler, U. Müller-Ladner (Hrsg.): Klinische Immunologie. Urban & Fischer, München 2012, S. 400.
- Kleonikos A. Tsakiris: Bibliography of Benediktos F. Adamantiades. In: Archiwum historii i filozofii medycyny. Band 79, Winter 2016, S. 8–15. (online)
- Ina Kötter: Über Morbus Behçet. Abgerufen am 8. Oktober 2017.