Sarkoidose
Die Sarkoidose (von altgriechisch σαρκωειδής sarkoeidés „fleischartig, fleischig“), auch als Morbus Boeck (buːk), Boecksche Krankheit oder Morbus Schaumann-Besnier bezeichnet, ist eine systemische Erkrankung des Bindegewebes mit Granulombildung, die meistens zwischen dem 20. und 40. Lebensjahr auftritt. Die genaue Ursache der Krankheit ist bis heute unbekannt.
| Klassifikation nach ICD-10 | |
|---|---|
| D86.0 | Sarkoidose der Lunge |
| D86.1 | Sarkoidose der Lymphknoten |
| D86.2 | Sarkoidose der Lunge mit Sarkoidose der Lymphknoten |
| D86.3 | Sarkoidose der Haut |
| D86.8 | Sarkoidose an sonstigen und kombinierten Lokalisationen |
| D86.9 | Sarkoidose, nicht näher bezeichnet |
| ICD-10 online (WHO-Version 2019) | |
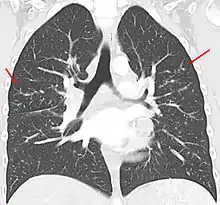
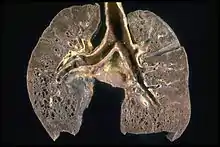
Bei der Sarkoidose bilden sich mikroskopisch kleine Knötchen (Granulome) in dem betroffenen Organgewebe, verbunden mit einer verstärkten Immunantwort.
Man unterscheidet eine zunächst akut verlaufende Form der Sarkoidose, das sogenannte Löfgren-Syndrom, von der schleichend und symptomarm einsetzenden chronischen Verlaufsform. In Deutschland tritt die Sarkoidose in 20 bis 30 Fällen auf 100.000 Einwohner auf.
Erstmals war sie von Ernest Besnier und Cæsar Peter Møller Boeck in den Jahren 1889 und 1899 als Hauterkrankung beschrieben worden. Im Jahre 1924 erkannte Jörgen Nilsen Schaumann, dass es sich hierbei um eine systemische Erkrankung verschiedener Organe handelt. Der Schwede Sven Halvar Löfgren beschrieb 1953 die nach ihm benannte akute Verlaufsform.
Epidemiologie
Sarkoidose ist eine weltweit vorkommende Krankheit, von der Männer etwas häufiger als Frauen betroffen sind, wobei sich die Zahlen mit zunehmendem Alter umkehren. Es gibt eine signifikante Häufung im Alter zwischen 20 und 30, wobei diese Häufung bei Männern noch ausgeprägter ist als bei Frauen. Die Fallzahlen nehmen nach dem dreißigsten Lebensjahr wieder deutlich ab. Kinder unter zehn Jahren sind praktisch nicht betroffen und ebenso Personen über 70.[1] In den USA und in (West-)Europa wird der Anteil der betroffenen Personen, die sogenannte Prävalenz, je nach Bemessungsgrundlage zwischen 1 und 40 pro 100.000 Einwohner angegeben. Eine überdurchschnittliche Häufung findet man bei Nordeuropäern und der afroamerikanischen Bevölkerung in den USA.[2] Den höchsten Anteil von Neuerkrankungen (Inzidenz) findet man in Schweden und Island mit über 60 Fällen pro 100.000 Einwohner pro Jahr. In Deutschland beträgt die Inzidenz 10–12 pro 100.000 Einwohner pro Jahr.[3] Die Epidemiologie legt eine genetische Mitursache nahe. In manchen Familien kommt Sarkoidose gehäuft vor, für nahe Verwandte eines Sarkoidosepatienten besteht eine circa zwanzigmal so hohe Wahrscheinlichkeit, ebenfalls zu erkranken, jedoch keine erhöhte Wahrscheinlichkeit für Ehepartner.[4]
Pathogenese
Eine erhöhte inflammatorische Aktivität und eine gesteigerte zelluläre Immunantwort mit Entstehung von nicht einschmelzenden Granulomen bilden die Pathogenese. Diese Granulome zeigen differenzierte Epitheloid- und Riesenzellen. Der beschriebenen Entzündungsreaktion liegt eine Störung der T-Lymphozytenfunktion bei gleichzeitig erhöhter B-Lymphozytenaktivität zugrunde. Dabei kommt es zu einer lokalen immunologischen Überaktivität mit oben beschriebener Granulombildung insbesondere im Lungengewebe und dem lymphatischen System. Besonders betroffen sind Lymphknoten (90 % der Fälle, Lymphknotensarkoidose) sowie die Lunge (90 %, Lungensarkoidose). Aber auch andere Organe wie Leber (60–90 %, Lebersarkoidose), Augen (25 %, Augensarkoidose), Herz (5 %, Herzsarkoidose), Skelett (25–50 %, Skelettsarkoidose), Milz (50–60 %, Milzsarkoidose) oder Haut (25–50 %, Hautsarkoidose) und das Knochenmark (15–40 %) können betroffen sein. Ist das Nervengewebe befallen, so spricht man von einer Neurosarkoidose. Da die Erkrankung familiär gehäuft auftreten kann, wird eine genetische Veranlagung vermutet. Im Februar 2005 wurde eine erste Genveränderung gefunden, die mit einem Ausbrechen der Krankheit korreliert. So reicht die Mutation eines einzigen Basenpaars im Gen BTNL2 auf Chromosom 6 aus, um die Erkrankungswahrscheinlichkeit um 60 % zu erhöhen. Eine Veränderung der Genkopien auf beiden Chromosomen erhöht das Risiko auf das Dreifache. BTNL2 beeinflusst eine Entzündungsreaktion, die bestimmte weiße Blutkörperchen aktiviert.
Symptome

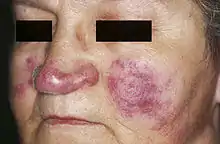
Meist ist die Lunge betroffen, da aber in etwa 30 % andere Organe betroffen sein können, sind die Symptome variabel. Meist zeigt sich die Erkrankung durch ein Druckgefühl im Oberkörper mit zunehmendem Husten bis hin zur Atemnot sowie durch Schwellung der Lymphknoten am häufigsten im Mediastinum. Die Patienten leiden häufig unter Müdigkeit (Fatigue) und Gelenkschmerzen. Beim häufigen Löfgren-Syndrom sind Fieber, Gelenkschmerzen, Leber- und Milzschwellungen sowie eine akute Entzündung des Unterhautfettgewebes mit „Knotenbildung“ (das Erythema nodosum[5]) meist an Unterschenkeln und Knöcheln die am häufigsten genannten Symptome. Bei Hautbeteiligung können auch Hautknötchen in unterschiedlichen Verteilungsmustern auftreten. Die an der Haut sichtbaren Veränderungen weisen als erstes wahrnehmbares Symptom oft auf die Diagnose hin. Eine charakteristische chronische Verlaufsform der Haut ist als Lupus pernio bekannt und imponiert durch eine bläuliche Schwellung mit Erosionen der Wangen, Nase, Lippen und Hände.
Bei Befall des Herzens können Herzrhythmusstörungen oder Herzmuskelschwäche auftreten. Selten findet man auch einen Pleuraerguss.[6] Bei einer Augenbeteiligung findet man häufig eine Uveitis (Entzündung der mittleren Augenhaut), die sich bei Beteiligung des Tränenganges zu einer Keratoconjunctivitis sicca ausweiten kann. Selten findet man bei einer Nierenbeteiligung einen gestörten Calciumstoffwechsel, welcher mit einer Nephrokalzinose (Nierenverkalkung) einhergehen kann. Die zystische Umwandlung der Fingerknochen (Jüngling-Syndrom) ist Ausdruck eines Knochenbefalls.
Eine Sonderform ist das Heerfordt-Syndrom, bei der es durch eine granulomatöse Entzündung der Ohrspeicheldrüsen zu einer Kompression des Gesichtsmuskelnerven und dadurch zu einer ein- oder beidseitigen teilweisen bis vollständigen Lähmung der Gesichtshälften (Fazialisparese) kommen kann.[7] Auch eine Beteiligung der Meningen (Hirnhäute) und der Nasennebenhöhlen mit möglicher späterer Zerstörung der Knorpelanteile wird beschrieben (Vorkommen bei 2 bis 18 % der Patienten). Sehr selten ist ein Befall des Hypothalamus-Hypophysen-Regelkreises mit einem daraus resultierenden Diabetes insipidus.
Diagnostik
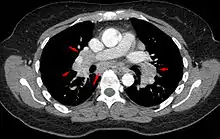
_lymph_node_biopsy.jpg.webp)
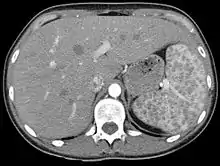
Da potenziell jedes Organ durch die Krankheit betroffen sein kann, richtet sich die Diagnostik nach der jeweiligen Beschwerdesymptomatik. Aufgrund der oft symptomlos verlaufenden Lymphknotenschwellungen wird die Sarkoidose nicht selten zufällig beim Röntgen-Thorax festgestellt. Je nach Befallsmuster und Zeichnung im Röntgenbild oder in der Computertomographie kann die pulmonale Sarkoidose in folgende Stadien (nach Scadding, 1967) eingeteilt werden, wobei diese Einteilung oft die Schwere und die Prognose der Erkrankung nicht wiedergibt:[8]
- Stadium 0: Normalbefund der Lunge bei Befall eines anderen Organs
- Stadium I: symmetrische Lymphknotenvergrößerung ohne sichtbaren Befall des Lungengewebes
- Stadium II: beidseitige Lymphknotenvergrößerung mit perilymphatischer Bildung von Granulomen im Lungengewebe
- Stadium III: Lungenbefall mit fehlender Lymphknotenvergrößerung
- Stadium IV: fibrotischer Umbau des Lungengewebes mit Funktionsverlust der Lunge.
Noch früher wurde die ältere Klassifikation nach Wurm (1958) mit den Stadien I bis III verwendet.
Im Labor finden sich bei der akuten Verlaufsform oft eine Erhöhung der Blutsenkungsgeschwindigkeit (BSG) sowie ein erhöhter Anteil der Blutkörperchen mit jüngerem Alter im Blutbild (die so genannte Linksverschiebung). Erhöhte Antikörper- und Immunglobulin-G-Werte bei mehr als der Hälfte der Patienten sind Zeichen einer erhöhten Aktivität der B-Zellen. Parallel dazu findet man eine Störung der T-Zellen, die sich klinisch in einem negativen Tuberkulin-Test äußern kann.
Auch das Angiotensin-Conversions-Enzym (ACE) und der lösliche Interleukin-2-Rezeptor (s-IL-2R) können bei über 60 % der Patienten erhöht sein, was aber auch bei anderen Erkrankungen vorkommt, so dass sie sich vorwiegend als Verlaufsparameter bzw. Aktivitätsparameter eignen. Bei Nierenbeteiligung misst man erhöhte Calcium-Mengen in Urin und Blut und einen erhöhten Calcitriol-Spiegel. Ein erhöhter Neopterin-Spiegel korreliert meist mit der Entzündungsaktivität der Makrophagen.
Eine Lungenfunktionsprüfung, meist auch eine Röntgenaufnahme der Lunge sowie eine Computertomographie werden durchgeführt, um die Therapiebedürftigkeit abschätzen zu können. Eine bronchoalveoläre Lavage (BAL) mit Zytologie kann oft diagnoseweisend sein. Sie zeigt typischerweise eine lymphozytäre Alveolitis mit Zunahme des CD4/CD8-Quotienten durch Erhöhung der Zahl der T-Helferzellen. Der Quotient beträgt beim Gesunden ca. 2, bei akuter Sarkoidose > 5. Eine transbronchiale Lungenbiopsie oder durch endobronchialen Ultraschall (EBUS) gezielte transbronchiale Biopsie der vergrößerten mediastinalen Lymphknoten kann histopathologisch zur Diagnosesicherung beitragen. Dabei werden in den Präparaten insbesondere nicht-verkäsende, epitheloidzellige Granulome mit Langhansschen Riesenzellen und einem Randwall aus Lymphozyten, Monozyten und Fibroblasten beobachtet. Das histologische Bild ist allerdings nicht spezifisch für die Sarkoidose. Die früher durchgeführte 67Ga-Szintigrafie wurde durch die FDG-PET/CT-Untersuchung abgelöst. Sie dient einerseits zur Kontrolle der Effektivität der durchgeführten Therapie, andererseits kann mit einer Untersuchung ein Überblick über die Anzahl der betroffenen Organsysteme gewonnen werden.
Differentialdiagnose
Die Diagnose erfolgt als Ausschlussdiagnose. Der Verdacht auf Sarkoidose muss vor allem von einer Lungentuberkulose, einer Tumoraussaat der Lunge (Lymphangiosis carcinomatosa) oder einem Lymphom abgegrenzt werden. Auch andere fibrosierende Lungenerkrankungen wie Langerhans-Zell-Histiozytose, exogen-allergische Alveolitis und Pneumokoniosen wie Silikose, Berylliose, Mischstaubsilikose oder Asbestose kommen in Frage. Bei Vorhandensein eines Erythema nodosum muss auch an Borreliose, Yersinien, diverse Bakterien, Mukoviszidose, Morbus Crohn, Lupus erythematodes und weitere Erkrankungen gedacht werden, die mit derselben Symptomatik einhergehen.
Verlauf und Prognose

Je nach Stadium sind die Verlaufsformen der Sarkoidose zu bewerten. Im Stadium I heilt die Sarkoidose bei einer akuten Verlaufsform fast immer ohne weiteren Therapiebedarf aus, wie auch im Stadium II eine hohe Spontanheilungsrate beobachtet wird. Insgesamt gesehen, kommt es bei etwa 60 % der Patienten in den ersten 3 Jahren zu einer spontanen Remission.[9] In anderen Fällen verläuft die Krankheit chronisch, meist schubförmig mit noch immer relativ günstiger Prognose. Nur wenige Fälle gehen in eine chronisch progrediente Verlaufsform über. Seltenere schwere Verlaufsformen können bis hin zur pulmonalen Hypertonie (mit cardialer Beteiligung als Cor pulmonale bezeichnet) und respiratorischer Insuffizienz (Atemnot) fortschreiten.
Als Faustregel gilt: Je jünger der Patient, je akuter der Verlauf, desto besser die Prognose.
Therapie Stand 2018
Eine kausale Therapie existiert bei der Sarkoidose nicht. Eine Therapie ist sinnvoll, wenn Beschwerden oder Komplikationen bestehen.[10] Da bis zu 60 % der Sarkoidosen eine Spontanremission haben, kann man zunächst engmaschig beobachten und versuchen, die unterschiedlichen Beschwerden symptomatisch zu lindern. Hingegen bei symptomatischem Organbefall wird eine Cortisontherapie durchgeführt, zum Beispiel bei funktionellen Einschränkungen der Lunge, davon insbesondere im Stadium III oder bei Hyperkalzämie (unter anderem bei Knochenmarksbefall). Die Dosierung beträgt oft 20–80 mg/Tag Prednisolon, nach einer Zeit kann versucht werden, zu reduzieren und dann auszuschleichen. Steroidbedingte Nebenwirkungen sollten möglichst gering gehalten werden. In manchen Fällen wird auch Methotrexat, meist in einer Dosierung von 10–15 mg/Woche, verwendet, um die Cortisondosis zu reduzieren.[11] In der Langzeittherapie können auch Immunsuppressiva wie Azathioprin und Chloroquin verwendet werden. Alle Therapien müssen engmaschig ärztlich überwacht werden.[12]
Beim Löfgren-Syndrom und in akuten Schüben werden statt (oder zusätzlich zu) Corticoiden auch Acetylsalicylsäure, Ibuprofen oder Diclofenac, gegebenenfalls auch zusätzlich Schmerzmittel eingesetzt. Wenn die Hauterscheinungen bei der Sarkoidose im Vordergrund stehen (kutane Sarkoidose), kann eine Therapie mit Tetracyclinen[13][14] oder Allopurinol versucht werden. Der Wirkmechanismus von Allopurinol ist hierbei ungeklärt.
Der Zeitpunkt des Beginns der Therapie mit Corticosteroiden bei Sarkoidose ist nicht unumstritten, da diese Therapie die Symptome nur unterdrückt und bei Beendigung der Therapie wieder auftreten lassen könnte. Eine Untersuchung zur Kortisontherapie[15] kam zu dem Ergebnis, dass die Behandlung mit Cortison die Rückfallquote wesentlich erhöhe. Als mögliche Ursache wird zum einen die Cortisonbehandlung an sich genannt, alternativ könnte es auch einfach darauf zurückzuführen sein, dass überwiegend schwerere Krankheitsverläufe mit Cortison behandelt wurden.[15] Kleine Studien geben Hinweise auf Wirkung des TNF-α-Blockers Infliximab[16] wie auch bei anderen Krankheitsbildern aus dem rheumatischen Formenkreis. Möglicherweise wird aber damit die Sterblichkeit erhöht.[17][18]
Eine begleitende psychotherapeutische Behandlung kann sich günstig auf den Krankheitsverlauf auswirken.[19]
Mögliche Mitursachen
Die Manifestationsorte Lunge, Haut und Augen lassen ein exogenes Agens erwarten, beispielsweise anorganische Staubbestandteile. So waren Feuerwehrleute vom Ground Zero häufiger von Granulomen betroffen.[20] Auch organische Stoffe, wie beispielsweise bakterielle DNA, wurden in Granulomen gefunden.
Gewisse Ähnlichkeiten zwischen Sarkoidose und Tuberkulose führten zum Verdacht, dass auch die Sarkoidose bakterielle Ursachen haben könnte. Versuche, die Sarkoidose mit Tuberkulosemedikamenten zu behandeln, blieben weitgehend erfolglos. In einer Metastudie wurden alle Arbeiten zwischen 1980 und 2006 zusammengefasst, die versuchten, Mykobakterien bei Sarkoidose mithilfe von PCR zu finden. Es ergab sich eine klare Assoziation von manchen Typen der Sarkoidose mit dem Vorhandensein der Erreger.[21] Andere Erreger als Ursache sind nicht ausgeschlossen.
In einigen Fällen ist die Auslösung einer Sarkoidose durch Interferon beschrieben, was zu einer relativen Kontraindikation dieses Medikaments bei Sarkoidose geführt hat.[22]
Geschichte
Als Erstbeschreiber der Sarkoidose (weitere Synonyme: Lymphogranuloma(tosis) benigna, Boeck-Besnier-Schaumannsche Krankheit, Lungentuberkuloid, benignes Miliarlupoid, multiples benignes Sarkoid, epitheloidzellige Reticuloendotheliose[23]) kann Jonathan Hutchinson (1828–1913) angesehen werden. Er stellte 1863 einen an Gicht erkrankten Patienten vor, der zusätzlich Hautveränderungen aufwies und vier Jahre später an Nierenversagen verstarb. Hutchinson führte dies jedoch auf die Gicht zurück – heute vermutet man, dass der aufgrund Sarkoidose veränderte Calciumstoffwechsel die eigentliche Ursache war. Der französische Dermatologe Ernest Henri Besnier (1831–1909) beschrieb 1889 eine symmetrische Hautveränderung der Extremitäten. Sein norwegischer Kollege Cæsar Peter Møller Boeck (1845–1917) erwähnte 1899 in dem wissenschaftlichen Aufsatz multiple benign sarcoid of the skin die histologischen Hautveränderungen und stellte schon damals den Verdacht einer systemischen Erkrankung. Die Hautveränderungen sind seitdem als Boecksche Sarkoidose bekannt.
Der dänische Ophthalmologe Christian Frederick Heerfordt (1871–1953) beschrieb 1909 eine fieberhafte Entzündung der Bindehaut mit Nervenbeteiligung und ordnete dies aufgrund der Laborwerte einem Mumps zu. Im Jahre 1924 bestätigte der schwedische Dermatologe Jörgen Nilsen Schaumann (1879–1953) die Entdeckungen Boecks, dass es sich hierbei um eine systemische Erkrankung verschiedener Organe handelt, und bezeichnete die Sarkoidose als Lymphogranulomatosis benigna, um sie vom Hodgkin-Lymphom abzugrenzen.
1941 beschrieb der norwegische Arzt Morten A. Kveim (1892–1966) den Kveim-Test zur Diagnostik der Sarkoidose, in den 1990er Jahren kam er allerdings außer Gebrauch.
Der Schwede Sven Halvar Löfgren (1910–1978) beschrieb 1953 die akute Verlaufsform anhand der Symptomtrias Erythema nodosum, Arthritis und bihiläre Lymphadenopathie (beidseitiger Befall der Lymphknoten in der Lunge). Dieses oft bei jungen Menschen anzutreffende Krankheitsbild wird als Löfgren-Syndrom bezeichnet.
Siehe auch
Literatur
Monographien
- Sarcoidosis. A Medical Dictionary, Bibliography, and Annotated Research Guide to Internet Reference. Icon Help Publications, San Diego CA 2004, ISBN 0-597-84072-5.
- James N. Parker (Hrsg.): The Official Patient’s Sourcebook on Sarcoidosis. Icon Help Publications, San Diego CA 2002, ISBN 0-597-83156-4.
- Karl Wurm: Sarkoidoseleitfaden. Thieme Verlag, Stuttgart 2000, ISBN 3-13-108012-4.
- Karl Wurm (Hrsg.): Sarkoidose. Thieme, Stuttgart 1983, ISBN 3-13-631701-7.
- Friedrich Wilhelm Bettinger (Hrsg.): Sarkoidoseleitfaden. Thieme Verlag, Stuttgart 1997, ISBN 3-13-108011-6 (Praxiswissen zu Klinik, Therapie und Prognose)
- R. Hoppe: Sarkoidose. Schattauer Verlag, Stuttgart 1965.
- J. Müller-Quernheim: Interstitielle Lungenerkrankungen. Thieme, 2003, ISBN 3-13-132281-0.
Leitlinien
- S1-Leitlinie Sarkoidose (im Kinder- und Jugendalter) der Deutschen Gesellschaft für Kinder- und Jugendmedizin (DGKJ). In: AWMF online (Stand 2013)
- S1-Leitlinie Chronische immunvermittelte ZNS-Erkrankungen der Deutschen Gesellschaft für Neurologie (DGN). In: AWMF online (Stand 2008)
- S1-Leitlinie Immunvermittelte Erkrankungen der grauen ZNS-Substanz sowie Neurosarkoidose der Deutschen Gesellschaft für Neurologie (DGN). In: AWMF online (Stand 2012)
Wissenschaftliche Arbeiten
- J. Hutchinson: On eruptions which occur in connection with gout. In: Jonathan Hutchinson (Hrsg.): Archives of Surgery. Mortimer’s malady, London 9.1898, S. 307, 315.
- E. Besnier: Lupus pernio de la face; synovites fongueuses (scrofulo-tuberculeuses) symétriques des extrémités superieures. In: Annales de Dermatologie et de Syphiligraphie. 2. Auflage. Masson, Paris 10.1889, S. 333–336.
- C. P. M. Boeck: Multiple benign sarcoid of the skin. In: Journal of cutaneous and genito-urinary diseases. Chicago-New York 17.1899, S. 543–550.
- E. Kuznitsky, A. Bittorf: Boecksches Sarkoid mit Beteiligung innerer Organe. In: Münchener Medizinische Wochenschrift. MMW Medizin-Verlag, München 62.1915, S. 1349–1353.
- J. Schaumann: Étude sur le lupus pernio et ses rapports avec les sarcoides et la tuberculose. In: Annales de dermatologie et de syphiligraphie. Masson, Paris 6.1916–1917, S. 357–373.
- F. R. Singer, J. S. Adams: Abnormal calcium homeostasis in sarcoidosis. In: The New England journal of medicine. Boston MA 315.1986,18 (12. Sep.), S. 755–757.
- T. Scharkoff: Epidemiologie der Sarkoidose. In: Pneumologie. Thieme, Stuttgart 47.1993,10 (Okt), S. 588–592.
- L. S. Newman, C. S. Rose, L. A. Maier: Sarcoidosis. In: The New England journal of medicine. Boston Mass 336.1997,17 (Apr 24), S. 1224–1234.
- R. Valentonyte et al.: Sarcoidosis is associated with a truncating splice site mutation in BTNL2. In: Nature Genetics. Advance online publication. New York 27. Februar 2005 (Bericht im IDW)
- M. Preiss: Laborchemische Veränderungen und Organbefall, insbesondere Blutbildveränderungen und Splenomegalie bei Sarkoidose. (PDF; 357 kB) Dissertation, Universität Marburg, 2003.
- A. Pfau, W. Stolz, S. Karrer, R.-M. Szeimies, M. Landthaler: Allopurinol in der Behandlung der kutanen Sarkoidose. In: Der Hautarzt. Band 49, Nr. 3/März 1998, S. 216–218.
- H. Nunes, D. Bouvry u. a.: Sarcoidosis. In: Orphanet Journal of Rare Diseases. Band 2, 2007, S. 46, doi:10.1186/1750-1172-2-46. PMID 18021432. PMC 2169207 (freier Volltext). (Review).
- C. Katic: Klinische Phänotypisierung des Verlaufs von 225 Sarkoidosepatienten. Dissertation. Albert-Ludwigs-Universität, Freiburg 2007.
Weblinks
Einzelnachweise
- Karl Wurm (Hrsg.): Sarkoidose. Thieme, Stuttgart/New York 1983, S. 12.
- L. S. Newman, C. S. Rose, L. A. Maier: Sarcoidosis. In: The New England Journal of Medicine. Band 336, Nummer 17, April 1997, S. 1224–1234, doi:10.1056/NEJM199704243361706. PMID 9110911. (Review).
- T. Scharkoff: Epidemiologie der Sarkoidose. In: Pneumologie. Band 47, Nummer 10, Oktober 1993, S. 588–592, PMID 8259367.
- Karl Wurm (Hrsg.): Sarkoidose. Thieme, Stuttgart / New York 1983, S. 13.
- Erythema nodosum associated with sarcoidosis. Bild eines Erythema nodosum bei MedlinePlus, abgerufen am 23. Mai 2012.
- Berthold Jany, Tobias Welte: Pleuraerguss des Erwachsenen – Ursachen, Diagnostik und Therapie. In: Deutsches Ärzteblatt. Band 116, Heft 21, (Mai) 2019, S. 377–385, hier: S. 379.
- Anisha Dua, Augustine Manadan: Heerfordt’s Syndrome, or Uveoparotid Fever. In: New England Journal of Medicine, Band 369, Ausgabe 5, 1. August 2013, S. 458, doi:10.1056/NEJMicm1303454.
- Piel, St. Diagnostik granulomatöser Erkrankungen mit Lungenbefall, in Pneumonews, 2017 9-(2) S. 40 ff.
- Sterling G. West: Current management of sarcoidosis I: pulmonary, cardiac, and neurologic manifestations. In: Current Opinion in Rheumatology. 30(3):243–248, Mai 2018, doi:10.1097/BOR.0000000000000489.
- S. Pabst, D. Skowasch, C. Grohé: Sarkoidose. In: Pneumologie. Band 66, Nummer 2, Februar 2012, S. 96–109, quiz 110, doi:10.1055/s-0030-1257126. PMID 22337329.
- R. P. Baughman, D. B. Winget, E. E. Lower: Methotrexate is steroid sparing in acute sarcoidosis: results of a double blind, randomized trial. In: Sarcoidosis, vasculitis, and diffuse lung diseases. Band 17, Nummer 1, März 2000, S. 60–66, PMID 10746262.
- C. S. King, W. Kelly: Treatment of sarcoidosis. In: Disease-a-month: DM. Band 55, Nummer 11, November 2009, S. 704–718, doi:10.1016/j.disamonth.2009.06.002. PMID 19857644. (Review).
- C. B. Doherty, T. Rosen: Evidence-based therapy for cutaneous sarcoidosis. In: Drugs. Band 68, Nummer 10, 2008, S. 1361–1383, PMID 18578557. (Review).
- A. N. Sapadin, R. Fleischmajer: Tetracyclines: nonantibiotic properties and their clinical implications. In: Journal of the American Academy of Dermatology. Band 54, Nummer 2, Februar 2006, S. 258–265, doi:10.1016/j.jaad.2005.10.004. PMID 16443056. (Review).
- J. E. Gottlieb, H. L. Israel u. a.: Outcome in sarcoidosis. The relationship of relapse to corticosteroid therapy. In: Chest. Band 111, Nummer 3, März 1997, S. 623–631, PMID 9118698.
- C. Barnabe, J. McMeekin u. a.: Successful treatment of cardiac sarcoidosis with infliximab. In: The Journal of Rheumatology. Band 35, Nummer 8, August 2008, S. 1686–1687, PMID 18671332.
- Eduard F. Stange: Morbus Crohn – Fragen zur Sicherheit und Effektivität von Infliximab. In: Deutsches Ärzteblatt. Jg. 102, Nr. 12, 25. März 2005, S. A826–A827 (aerzteblatt.de [PDF; abgerufen am 23. März 2009]).
- A. Rothova: Ocular involvement in sarcoidosis. In: The British journal of ophthalmology. Band 84, Nummer 1, Januar 2000, S. 110–116, PMID 10611110. PMC 1723211 (freier Volltext). (Review).
- G. Trombini, E. Trombini: Sarcoidosis: Psychotherapy and Long-Term Outcome – A Case Report. In: Case Reports in Medicine. Vol. 2012, Article ID 232491.
- G. Izbicki, R. Chavko u. a.: World Trade Center “sarcoid-like” granulomatous pulmonary disease in New York City Fire Department rescue workers. In: Chest. Band 131, Nummer 5, Mai 2007, S. 1414–1423, doi:10.1378/chest.06-2114. PMID 17400664.
- D. Gupta, R. Agarwal u. a.: Molecular evidence for the role of mycobacteria in sarcoidosis: a meta-analysis. In: The European respiratory journal. Band 30, Nummer 3, September 2007, S. 508–516, doi:10.1183/09031936.00002607. PMID 17537780.
- Wolff H. Schmiegel: Diagnostik und Therapie der akuten und chronischen Hepatitis C – Vorgehen in Problemsituationen. In: Zeitschrift für Gastroenterologie. Band 42, Nr. 8. Georg Thieme, Stuttgart / New York 11. August 2004, S. 720–723, doi:10.1055/s-2004-813445, PMID 15314723 (dgvs.de [PDF; abgerufen am 27. Oktober 2012]).
- Joachim Frey: Boecksche Krankheit. In: Ludwig Heilmeyer (Hrsg.): Lehrbuch der Inneren Medizin. Springer-Verlag, Berlin/Göttingen/Heidelberg 1955; 2. Auflage ebenda 1961, S. 705–708, hier: S. 705.