Amyloidose
Die Amyloidose (von altgriechisch ἄμυλον ámylon „Kraftmehl, Stärke“)[1] ist eine variable Erkrankung mit Anreicherung von (zum Teil abnorm veränderten) Proteinen meist extrazellulär im Interstitium (Zwischenzellraum). Diese unlöslichen Ablagerungen liegen in Form kleiner Fasern, so genannter Fibrillen (β-Fibrillen), vor und werden als Amyloid bezeichnet. Der Nachweis von Amyloid erfolgt durch mikroskopische Untersuchung von Gewebeproben, die zuvor mit Kongorot gefärbt wurden. Die Amyloidablagerungen erscheinen im Hellfeld hellrot, in polarisiertem Licht zeigen sie eine apfelgrüne Doppelbrechung.
| Klassifikation nach ICD-10 | |
|---|---|
| E85 | Amyloidose |
| ICD-10 online (WHO-Version 2019) | |
Die Ablagerung von Corpora amylacea in der Prostata gilt als einzige nicht krankhafte Form der Amyloidablagerung im Körper. Amyloidose selbst ist keine einzelne Krankheit, sondern ein krankhafter Ablagerungsprozess (durch die genannten fibrillären Proteinablagerungen), der von unterschiedlichen Stoffwechseldefekten ausgelöst wird und – je nach betroffenem Organ – zu verschiedenen chronischen Erkrankungen führen kann.
Amyloid bedeutet so viel wie „stärkeähnlich“, denn die Ablagerungen zeigen häufig bei Zugabe von Iod eine Blaufärbung, ähnlich wie die Iod-Stärke-Reaktion. Rudolf Virchow benutzte den Begriff 1854 zum ersten Mal, als er atypisches Material in der Leber von Verstorbenen fand.
Die Inzidenz der systemischen Amyloidose (die den ganzen Körper betreffende Amyloidose als Multisystemerkrankung) beträgt etwa 1 Fall pro 100.000 Einwohner und Jahr. Betroffen sind vorwiegend ältere Patienten um 65 Jahre. Die systemische Erkrankung ist auch mit intensiver Behandlung (Zytostatika, Glucocorticoide) häufig tödlich; nur manchmal – abhängig von der biochemischen Ursache – ist eine Behandlung der Ursache möglich.
Als Hauptformen der Amyloidose gelten heute die Leichtketten-Amyloidose (AL) und die Transthyretin-bedingte ATTR-Amyloidose.
Bei der unbehandelten AA-Amyloidose (eine Form der Amyloidose, bei der die unlöslichen Fibrillen aus Amyloid A bestehen, dessen Vorläuferprotein ein Akute-Phase-Protein ist) beträgt die mediane Überlebenszeit 3 bis 4 Jahre[2]. Ausheilungen sind möglich, wenn die Ursache eine Infektion war. Unter allen systemischen Amyloidose-Formen ist die AL-Amyloidose am häufigsten anzutreffen.

Ursache und Entstehung
Der Amyloidose liegt eine Störung der Faltung eines normalerweise löslichen Proteins zu Grunde.[3] Mehrere verschiedene Krankheiten können durch Überproduktion, fehlenden/verminderten Abbau oder gestörte Ausscheidung bestimmter Proteine die Erkrankung auslösen. Diese Proteine liegen üblicherweise im Blutplasma in gelöster Form vor. Steigt ihre Konzentration aber an, so gelangen sie auch in das umgebende Gewebe, wo Enzyme sie angreifen. Durch Zusammenlagerung der entstehenden Aminosäureketten im Bereich der β-Faltblatt-Strukturen bilden sich unlösliche Komplexe in Form mikroskopisch kleiner Fasern (Fibrillen).
Da die Fibrillen gegenüber den Abwehrmechanismen (Phagozytose und Proteolyse) resistent sind, können sie nicht mehr entfernt werden. Neueren Erkenntnissen zufolge können allerdings durch eine Entfernung der Vorläuferproteine die Amyloid-Ablagerungen zumindest reduziert werden.
Die Amyloidablagerungen zerstören die Architektur der Organe und führen dadurch zu Funktionsstörungen. Es gibt Hinweise, dass die Ablagerungen auch einen direkten toxischen Effekt auf Zellen ausüben.
Diagnose
Die Symptome der Amyloidose sind vor allem in frühen Stadien der Erkrankung oft unspezifisch. Aus diesem Grund wird die Diagnose oft erst in einem fortgeschrittenen Krankheitsstadium gestellt. An eine Amyloidose sollte gedacht werden, wenn eine anders nicht erklärte Proteinurie vorliegt, des Weiteren bei Patienten mit Kardiomyopathie, Neuropathie, Lebervergrößerung oder bei Vorliegen eines multiplen Myeloms.
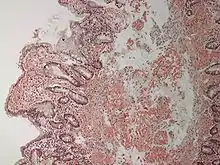
Da, wenn nicht ein szintigrafischer Nachweis einer kardialen ATTR-Amyloidose (Transthyretin-Amyloidose) bei Ausschluss einer monoklonalen Gammopathie erfolgt ist, die genaue Zusammensetzung des Amyloids für die Behandlung bekannt sein muss, ist meist eine Biopsie (Gewebeprobe) zum histologischen Amyloidnachweis mit Subtypisierung erforderlich. Diese kann aus einem befallenen Organ (Niere, Herz, Magen) gewonnen werden. Eine neue Entdeckung ist, dass generalisierte Amyloidosen auch mit Biopsien aus dem Unterhaut-Fettgewebe eingeordnet werden können, eine für den Patienten weniger belastende Prozedur. Weitere wenig belastende Entnahmestellen sind die kleinen Speicheldrüsen, das Zahnfleisch, der Enddarm oder die Haut. Die histologisch aufbereiteten Proben werden mit Kongorot gefärbt und immunhistochemisch untersucht. Amyloid bindet den Farbstoff Kongorot und wird dann unter polarisiertem Licht grünlich leuchtend sichtbar. Das Elektronenmikroskop zeigt, dass die Amyloidablagerungen aus Fibrillen bestehen, irregulär angeordneten, fadenförmigen Strukturen unterschiedlicher Länge mit einem Durchmesser zwischen 8 und 15 nm.
Wurde durch Gewebeentnahme eine Amyloidose gesichert, sollte durch Knochenmarkpunktion, Blut- und Urinuntersuchung nach einer Leichtketten-Erkrankung gesucht werden. Im Knochenmark spricht eine monoklonale Vermehrung der Plasmazellen, d. h. von Plasmazellen, die entweder nur kappa- oder nur lambda-Ketten produzieren, für eine AL-Amyloidose. Die Leichtketten können auch durch eine Immunelektrophorese in Serum oder Urin nachgewiesen werden.
Das Ausmaß der Amyloidablagerungen kann durch Szintigraphie mit radioaktiv markierten Substanzen, die an Amyloid binden, sichtbar gemacht werden. An Amyloid binden Technetium Tc 99m-Pyrophosphat, Technetium-markiertes Aprotinin und I-markierte Serum-Amyloid P-Komponente. Das Ausmaß der Herzbeteiligung kann durch Echokardiografie oder Kernspintomografie sichtbar gemacht werden.

In vielen Fällen wird die Diagnose erst nach dem Tod durch die Autopsie gestellt. Eine übergroße Zunge und charakteristische derbe Unterhautschwellungen weisen den Pathologen auf die richtige Diagnose. Aufgrund des speckartigen Aussehens der Schnittflächen befallener Organe werden diese auch als Speckleber, Speckmilz oder Speckniere bezeichnet. Genau wie Virchow kann er die Schnittfläche eines beliebigen Organs (so auch Zunge, Herz) mit Lugolscher Lösung beträufeln, welche mit Amyloid eine charakteristische Braunfärbung ergibt.
Einteilung
Früher wurden die Amyloidosen wie folgt eingeteilt:
| Typ | Häufigkeit | Details |
|---|---|---|
| Primäre Amyloidose | selten | keine Assoziation zu anderen Grunderkrankungen Ursache unbekannt meist Ablagerungen vom sogenannten AL-Typ (s. u.) |
| Familiäre Amyloidose | selten | tritt gehäuft in Kombination mit vererbten Erkrankungen auf z. B. familiäres Mittelmeerfieber meist Ablagerungen vom AA-Typ v. a. Niere betroffen |
| Sekundäre Amyloidose | am häufigsten | ausschließlich bei anderen Grunderkrankungen auftretend z. B. chronische infektiöse und nichtinfektiöse Entzündungen, Tumoren des lymphatischen Systems, längere Dialyse Ablagerungen verschiedenster Amyloid-Subtypen |
| Altersamyloidose | häufig | im Alter ohne zugrundeliegende Erkrankung auftretend Ablagerungen v. a. von AS-Amyloid hauptsächlich Herz und Gehirn betroffen |
Bis heute wurden in Amyloidablagerungen zehn verschiedene Proteine nachgewiesen (Jahr des Nachweises in Klammern):
- Immunglobulin-Leichtketten (1971)
- Amyloid A (1972)
- Transthyretin (1981)
- Cystatin C (1986)
- Apolipoprotein A-I (1988)
- Gelsolin (1990)
- Fibrinogen Aα-Kette (1993)
- Lysozym (1993)
- Apolipoprotein A-II (2001)
- Leukozyten Chemotaktischer Faktor 2, LECT2 (2008)[4]
Die Amyloidosen werden entsprechend dem beteiligten Protein eingeteilt, welches immunhistochemisch differenzierbar ist. Bei der Benennung steht das erste A für „Amyloidprotein“; der zweite Buchstabe bezeichnet den Typ des betroffenen Proteins.
AA-Amyloidose
Diese Form wird durch Serum-Amyloid-A, ein sogenanntes Akute-Phase-Protein, verursacht. Zu einer vermehrten Bildung des Amyloid-A kommt es beim autosomal-rezessiv vererbten familiären Mittelmeerfieber. Ferner rufen chronische Infektionen (wie Tuberkulose, Lepra oder Osteomyelitis) und chronische nichtinfektiöse Entzündungen (z. B. Colitis ulcerosa, Morbus Crohn, Psoriasis, Morbus Bechterew, Kollagenosen, rheumatoide Arthritis), sowie auch Tumoren (z. B. Morbus Hodgkin, Karzinome) manchmal diesen Effekt hervor. Die Symptome der AA-Amyloidose treten üblicherweise generalisiert im Körper auf. Besonders betroffen sind Niere, Milz, Leber, Nebennieren und immer der Magen-Darm-Trakt.
AL-Amyloidose
Die AL-Amyloidose (Amyloid, bestehend aus Leichtketten) ist die häufigste der systemischen Amyloidosen. Es handelt sich dabei um eine Erkrankung der Plasmazellen, also derjenigen Zellen im Knochenmark, die beim Gesunden für die Produktion der Antikörper (Immunglobuline) verantwortlich sind. Ein Antikörper besteht jeweils aus zwei schweren und zwei leichten Ketten. Bei der Immunglobulin-Synthese in den Plasmazellen werden die Leichtketten immer im Überschuss gebildet, so dass neben den intakten Antikörpern auch freie Leichtketten in das Blut gelangen. Bei der AL-Amyloidose kommt es zu einer pathologischen Vermehrung der Plasmazellen, und dadurch zur Bildung von monoklonalen freien Leichtketten oder deren Fragmenten. Diese strukturell veränderten Leichtketten werden als Amyloid abgelagert. Lambda-Leichtketten werden etwa dreimal häufiger in den Ablagerungen gefunden als Kappa-Leichtketten. Oftmals kann man die gleichen Proteine als sogenannte Bence-Jones-Proteine auch im Harn nachweisen. Für die Überproduktion ist meist ein Tumor des lymphatischen Gewebes (Lymphom) verantwortlich. Sehr häufig kommt sie beim multiplen Myelom (bis zu 15 %) und dem Morbus Waldenström vor. Die AL-Amyloidose entspricht der früheren „primären Amyloidose“. Sie tritt meist lokalisiert in einzelnen Organen auf. Am häufigsten betroffen sind Herz und Nieren, mit Ausnahme des Gehirns kann aber jedes andere Organ befallen sein.
AE-Amyloidose
Hier sind Proteinhormone aus endokrinen (nach innen ausscheidenden) Drüsen betroffen; z. B. Insulin oder Calcitonin. Sie stammen aus hormonproduzierenden Tumoren, etwa bestimmten Tumoren der Bauchspeicheldrüse oder der Schilddrüse. Die Symptome liegen lokalisiert vor und betreffen häufig Nerven, Herz, Niere, Drüsen, Lunge oder Haut.
AB-Amyloidose
Sie leitet sich vom Beta-2-Mikroglobulin ab, einem Protein, das sich unter langjähriger Dialyse anhäuft. AB-Amyloidose führt typischerweise zu einem Karpaltunnelsyndrom sowie zu schmerzhaften Ablagerungen in Gelenken.
AP-Amyloidose
Ablagerungen von Präalbumin (→ Transthyretin), die Hauptlokalisation liegt im Nerven- und Herzgewebe.
AS-Amyloidose
Hier werden verschiedene Proteine zusammengefasst, die eine senile Amyloidose zur Folge haben. Vielfach sind die auslösenden Proteine noch nicht charakterisiert. Ein gehäuftes Auftreten wird mit der Alzheimerschen Krankheit in Verbindung gebracht.
ATTR-Amyloidose
Transthyretin (TTR) ist ein Serumprotein, welches Thyroxin und Retinol transportiert.[5] Bei Entzündungszuständen ist der Serumspiegel vermindert. Deshalb wird es auch als Anti-Akute-Phase-Protein bezeichnet. TTR hat die Fähigkeit, zu unlöslichen Aggregaten zu akkumulieren, welche Zellen und Gewebe schädigen können. Einzelne Punktmutationen können die Fähigkeit steigern, durch fehlerhafte Faltung unlösliche Ablagerungen von Aggregaten in Herz, Nerven und Leptomeninx (weiche Hirnhaut) zu bilden.
Auch nicht mutiertes TTR kann durch Fehlfaltung eine Amylodkonfiguration erhalten (wild-type TTR-Amyloidose, wt-TRR-Amyloidose). Da diese Form sporadisch bei Männern über 60 Jahre auftritt, wurde sie früher "senile kardiale Amyloidose" oder "senile systemische Amyloidose" genannt.[5] Darüber hinaus können genetisch veränderte Varianten des Transthyretin zu vererblichen Amyloidosen mit autosomal-dominantem Erbgang führen (hereditäre ATTR-Amyloidose, ATTRv-Amyloidose).[6] Bislang sind mehr als 80 Mutationen bekannt. Die Ablagerungen erfolgen in Augen, Nieren und Herz und selten im zentralen Nervensystem. Zur Behandlung der ATTR-Amyloidose können die Medikamente Patisiran[7] und Tafamidis[8] eingesetzt werden.
Apo A1 Amyloidose
Ebenfalls autosomal dominanter Erbgang, welcher sich jedoch bereits in jungen Jahren der Patienten bemerkbar machen kann. Häufig betroffene Organe sind die Niere, das Herz, Magen-Darm-Trakt, sowie Polyneuropathien und autonome Neuropathien. Für diese Form der Amyloidose gibt es keine Therapiemöglichkeit.
Symptome
Herz
Die Ablagerung der Proteinfibrillen um die Muskelfasern des Myokards verursachen eine Versteifung der Herzmuskulatur (restriktive Kardiomyopathie). Dies führt über eine rasche Verschlechterung der Pumpleistung des Herzens zur Herzschwäche (Herzinsuffizienz, auch zur Herzinsuffizienz bei erhaltener Pumpleistung). Durch die Ablagerungen sind die Herzwände verdickt bei normaler oder verminderter Größe der Herzkammern. Die Herzschwäche kann verbunden sein mit Dyspnoe, peripheren Ödemen, Anasarka, Pleuraerguss, Perikarderguss und Hypotension.[9] Häufig ist im EKG die Höhe der QRS-Komplexe vermindert (Niedervoltage). Wenn das Erregungsleitungssystem des Herzens betroffen ist, können Herzrhythmusstörungen die Folge sein. Bei einem Befall der Herzkranzgefäße können Durchblutungsstörungen entstehen.[10] Bei Plasmazelldyskrasie findet sich eine unerklärte kardiale Biomarkererhöhung.
Niere
Die krankhaften Proteine können sich um zu- und ableitende Gefäße (vaskulär betonte Amyloidose) oder um die Kapillargefäße des Nierenkörperchens (glomerulär betonte Amyloidose) ansammeln. Die Folge ist in den meisten Fällen eine stark erhöhte Eiweißausscheidung (mit Ödemen, schäumendem Urin bis zum nephrotischen Syndrom) und ein fortschreitender Nierenfunktionsverlust. Bei der vaskulär betonten Amyloidose (etwa 10 % der Fälle) kann es auch zum Nierenfunktionsverlust kommen, ohne dass eine erhöhte Eiweißausscheidung im Urin nachweisbar ist.[11] Wird aufgrund einer Amyloidose eine Dialysebehandlung erforderlich, liegt die mittlere Lebenserwartung bei AL-Amyloidose bei 26 Monaten, bei AA-Amyloidose ist die Prognose deutlich besser. Entscheidend für die Prognose ist die Herzbeteiligung.[12]
Verdauungstrakt
Die Folgen der Amyloidose im Verdauungstrakt können hier eine Makroglossie (Vergrößerung der Zunge, Kennzeichen der AL-Amyloidose), Schluckbeschwerden (Dysphagie), Appetitlosigkeit, Fatigue und mangelnde Aufnahme von Nahrungsbestandteilen (Malabsorption) mit Gewichtsverlust, Störungen in der Beweglichkeit des Darmes mit Übelkeit, Völlegefühl, Meteorismus, Durchfällen, Obstipation, gastrointestinale Blutungen, Perforationen und Darmverschluss sein. Häufig ist die Leber vergrößert (unerklärte Hepatomegalie) und in Folge einer erhöhten Lebersteifigkeit beim Abtasten steinhart. Zudem kann es zur Ausbildung eines Aszites kommen. Bei der Blutuntersuchung fällt meist eine deutliche Erhöhung der alkalischen Phosphatase bei nur moderater Erhöhung der Transaminasen auf.
Gehirn und Nerven
Am Gehirn können Funktionsstörungen aller Art bis hin zur Demenz entstehen (vgl. Alzheimersche Krankheit). An den peripheren Nerven kommt es zu oft sehr schmerzhaften Gefühls- oder Bewegungsstörungen. Ein Befall des vegetativen Nervensystems führt durch (orthostatische) Dysregulation zu Blutdruckabfall im Stehen (Orthostase-Reaktion), verfrühtem Sättigungsgefühl infolge verminderter Magenentleerung, Erektionsstörungen, Blasenentleerungsstörung und Störungen der Darm-Beweglichkeit (Darmmotilitätsstörung) mit Blähungen, Bauchschmerzen und Stuhlunregelmäßigkeiten. Symptome des Befalls von peripherem und autonomem Nervensystem können sich als variables Bild einer (rasch progredienten) Polyneuropathie zeigen. Auch eine lumbale Spinalkanalstenose kann eine Manifestationsform sein.
Weichteilgewebe, Auge
Weichteilbeteiligung äußert sich in Karpaltunnelsyndrom, Bizepssehnenruptur, Knoten im Bereich der Haut, Erkrankungen der Gelenke (Arthropathie mit Gelenkschwellungen), Muskelschwäche, Haarausfall (Alopezie), Veränderungen im Bereich von Finger- und Zehennägeln (Nageldystrophie), Vergrößerung der Unterkieferspeicheldrüse, trockene Augen, geröteten Augenringen bzw. periorbitalen Hauteinblutungen (Purpura durch Gerinnungsstörungen), Glaskörpertrübung, Grüne Star, retinaler Angiopathie und Heiserkeit.
Endokrines System
In seltenen Fällen können bei der AL-Amyloidose infolge von Amyloid-Ablagerungen Störungen im endokrinen System auftreten, die sich bei den hormonproduzierenden Drüsen als Schilddrüsen- oder Nebennierenunterfunktion zeigen.
Lunge
Durch Bildung von Exsudat kann es zu einem Pleuraerguss kommen.[13]
Milz
Auch eine Splenomegalie (Vergrößerung der Milz) kann Symptom der systemischen Amyloidose sein.
Therapie
Allgemeinmaßnahmen
- Herzbeteiligung: Die Herzschwäche wird behandelt mit kochsalzarmer Kost, Diuretika und ACE-Hemmern. Bei Auftreten von Ohnmachtsanfällen kann ein Herzschrittmacher erforderlich werden. Ventrikuläre Herzrhythmusstörungen werden mit Amiodaron behandelt; unter Umständen wird ein implantierbarer Kardioverter-Defibrillator erforderlich. Ein ausgeprägter Blutdruckabfall im Stehen ist sehr schwierig zu behandeln, zum Einsatz kommen Kompressionsstrümpfe, Midodrin, in schweren Fällen Norepinephrin-Infusionen.
- Nierenbeteiligung: Die Behandlung erfolgt mit kochsalzarmer Kost, Diuretika, medikamentöser Senkung erhöhter Blutfette, ACE-Hemmern und AT1-Antagonisten. Sinkt die Nierenfunktion unter 15 % der Norm, wird eine Dialysebehandlung erforderlich.
- Durchfälle aufgrund einer Schädigung des vegetativen Nervensystems sprechen häufig auf Octreotid an. Neuropathische Schmerzen sind auch mit Gabapentin nur schwer zu beeinflussen.
AA-Amyloidose
- Bei Patienten mit familiärem Mittelmeerfieber kann die lebenslange Einnahme von Colchicin die Entstehung einer Amyloidose verhindern.
- Experimentelle Therapien sind die Gabe des TNF-Blockers Etanercept und von 1,3-Propandisulfonat, welches die Fibrillenbildung verhindern und Fibrillenablagerungen auflösen soll.
- Eprodisat ist ein negativ geladenes, niedermolekulares sulfoniertes Molekül, das strukturelle Ähnlichkeiten zu Heparansulfat aufweist. Eprodisat hemmt die Interaktion zwischen Amyloid bildenden Proteinen und Glykosaminoglykanen. Im Maus-Modell hemmt Eprodisat die Ablagerung von AA-Amyloid. In einer randomisierten, placebokontrollierten Doppelblindstudie verlangsamte Eprodisat bei Patienten mit AA-Amyloidose die Verschlechterung der Nierenfunktion.[14]
AL-Amyloidose
Ziel der Behandlung ist, durch Hemmung der Vermehrung von Plasmazellen die Produktion der abnormalen Leichtketten zu vermindern.
- Die wirksamste Behandlung ist eine hochdosierte Chemotherapie mit Melphalan mit anschließender Übertragung eigener Blutstammzellen (autologe Stammzelltransplantation). Die Sterblichkeit unter Behandlung liegt bei 15–40 %, das Risiko ist erhöht bei fortgeschrittener Herzbeteiligung, verminderter Nierenfunktion, Beteiligung von mehr als zwei Organen, schwerer Hypotension und schlechtem Allgemeinzustand. Eine vorangegangene konventionelle Chemotherapie mit Melphalan vermindert die Anzahl der zur Verfügung stehenden Stammzellen, dies sollte bei der Therapie-Planung berücksichtigt werden.
- Die Übertragung fremder Knochenmarkzellen (allogene Knochenmarktransplantation) ist eine experimentelle Behandlung und wurde bislang nur in wenigen Fällen durchgeführt. Die Sterblichkeit liegt bei 40 %.
- Konventionelle Chemotherapie erfolgt durch orale Gabe von niedrig dosiertem Melphalan und Prednison über jeweils eine Woche in Abständen von 6 Wochen. Der Effekt auf die Überlebensdauer ist moderat, im Median überleben die Patienten unter dieser Therapie etwa 18 Monate. Eine konventionelle Chemotherapie vermindert die Anzahl der Stammzellen im Knochenmark und schmälert so die Erfolgsaussichten einer späteren Stammzelltransplantation. Deshalb ist vor Beginn einer konventionellen Chemotherapie immer zu prüfen, ob der Patient für eine Stammzelltransplantation in Frage kommt.
- Spricht die Behandlung nicht an, oder kommt es zu einem Rückfall, kann ein Behandlungsversuch mit Thalidomid in Kombination mit Dexamethason unternommen werden. Thalidomid, das unter dem Handelsnamen Contergan zu trauriger Berühmtheit gelangt ist, darf wegen der Gefahr der Fruchtschädigung von Frauen im gebärfähigen Alter nur unter Einhaltung besonderer Sicherheitsmaßnahmen eingenommen werden.
- Experimentelle Therapien sind die Gabe des Proteasom-Inhibitors Bortezomib und des Thalidomid-Analogons Lenalidomid. Im Tierversuch bindet 4-Iodo-4-Deoxy-Doxorubicin an Amyloid-Fibrillen und vermag diese aufzulösen, Studien am Menschen verliefen bislang allerdings enttäuschend. Der TNF-α-Inhibitor Etanercept kann bei schwerer AL-Amyloidose die Beschwerden lindern, hat aber keinen Einfluss auf die zugrunde liegende Plasmazell-Erkrankung. Im Tierversuch kann die Amyloid-Beladung vermindert werden durch eine passive Immunisierung mit einem monoklonalen Antikörper gegen ein Epitop, das spezifisch ist für Amyloid-bildende Leichtketten.
- Die Mediziner Giampaolo Merlini und Giovanni Palladini starteten 2012 in Pavia, Italien, die erste randomisierte klinische Studie über Epigallocatechingallat bei kardialer AL-Amyloidose.[15]
ATTR-Amyloidose
- Die Behandlung erfolgte bislang vor allem durch Leber- und in Ausnahmefällen Herztransplantation.[16] Weil bis zum Auftreten von Symptomen 20 bis 30 Jahre vergehen, kann die kranke Leber eventuell im Rahmen einer Dominotransplantation einem anderen Patienten wieder eingesetzt werden.[17]
- In einer Studie an der Medizinischen Klinik der Universität Heidelberg seit April 2008 über die Behandlung von Patienten mit Transthyretin-Amyloidose mit Epigallocatechingallat zeigten sich bei einem Teil der Patienten Verbesserungen bei der Herzfunktion.[18] Der Ansatz von Werner Hunstein und anderen wird durch andere Untersuchungsergebnisse unterstützt.[19]
- Patisiran (Handelsname: Onpattro, Alnylam Pharmaceuticals) wurde in den USA im August 2018 von der FDA zugelassen[20], und im gleichen Monat folgte in der Europäischen Union die Zulassung zur Behandlung der hATTR-Amyloidose bei erwachsenen Patienten mit Polyneuropathie der Stadien 1 oder 2.[21]
Literatur
- Christoph Röcken u. a.: Interdisziplinäre Leitlinien zur Diagnostik und Therapie der extrazerebralen Amyloidosen. In: Deutsche Gesellschaft für Amyloid-Krankheiten e. V. (Hrsg.): Deutsche Medizinische Wochenschrift. Band 131, Suppl 2, 2006, S. 45–66, doi:10.1055/s-2006-947836.
- Sandra Ihne, Caroline Morbach, Claudia Sommer, Andreas Geier, Stefan Knop, Stefan Störk: Amyloidose – Diagnostik und Therapie einer unterdiagnostizierten Erkrankung. In: Deutsches Ärzteblatt. Band 117, Nr. 10, 6. März 2020, S. 159–166.
Weblinks
- Amyloidose Pathologie – Bilddatenbank Pathopic der Universität Basel (PathoPic – Anleitung; PDF; 2,2 MB)
- Wikilite.com Englischsprachige Seite mit umfangreichen Informationen zu freien Leichtketten und zur Leichtketten-Amyloidose (AL-Amyloidose)
Einzelnachweise
- Wilhelm Pape, Max Sengebusch (Bearb.): Handwörterbuch der griechischen Sprache. 3. Auflage, 6. Abdruck, Vieweg & Sohn, Braunschweig 1914. 1914, abgerufen am 9. Dezember 2015.
- Bouke P.C. Hazenberg: Amyloidosis: a clinical overview. Band 39, Nr. 2, Mai 2013, S. 323–345, doi:10.1016/j.rdc.2013.02.012 (rug.nl [PDF; abgerufen am 8. September 2019]).
- G Merlini, V Bellotti: Molecular mechanisms of amyloidosis. In: N Engl J Med. Band 349, Nr. 6, 2003, S. 583–596, doi:10.1056/NEJMra023144.
- Merrill D Benson: LECT2 amyloidosis. In: Kidney International. Band 77, Nr. 9, Mai 2010, S. 757–759, doi:10.1038/ki.2010.18, PMID 20393490.
- Morie A. Gertz, Merrill D. Benson, Peter J. Dyck, Martha Grogan, Terresa Coelho: Diagnosis, Prognosis, and Therapy of Transthyretin Amyloidosis. In: Journal of the American College of Cardiology. Band 66, Nr. 21, Dezember 2015, S. 2451–2466, doi:10.1016/j.jacc.2015.09.075 (elsevier.com [abgerufen am 30. Oktober 2021]).
- Yukio Ando, Teresa Coelho, John L Berk, Márcia Waddington Cruz, Bo-Göran Ericzon: Guideline of transthyretin-related hereditary amyloidosis for clinicians. In: Orphanet Journal of Rare Diseases. Band 8, 20. Februar 2013, ISSN 1750-1172, S. 31, doi:10.1186/1750-1172-8-31, PMID 23425518, PMC 3584981 (freier Volltext).
- David Adams, Alejandra Gonzalez-Duarte, William D. O’Riordan, Chih-Chao Yang, Mitsuharu Ueda: Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. In: New England Journal of Medicine. Band 379, Nr. 1, 5. Juli 2018, ISSN 0028-4793, S. 11–21, doi:10.1056/NEJMoa1716153 (nejm.org [abgerufen am 30. Oktober 2021]).
- Mathew S. Maurer, Jeffrey H. Schwartz, Balarama Gundapaneni, Perry M. Elliott, Giampaolo Merlini: Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. In: New England Journal of Medicine. Band 379, Nr. 11, 13. September 2018, ISSN 0028-4793, S. 1007–1016, doi:10.1056/NEJMoa1805689 (nejm.org [abgerufen am 30. Oktober 2021]).
- Sandra Ihne, Caroline Morbach, Claudia Sommer u. a.: Amyloidose – Diagnostik und Therapie einer unterdiagnostizierten Erkrankung. 2020, S. 160 und 165.
- Keyur B. Shah u. a.: Amyloidosis and the Heart: A Comprehensive Review. In: Arch Intern Med. Band 166, Nr. 17, 2006, S. 1805–1813, doi:10.1001/archinte.166.17.1805.
- Vaishali Sanchorawala: Light-Chain (AL) Amyloidosis: Diagnosis and Treatment. In: Clin J Am Soc Nephrol. Band 1, Nr. 6, 2006, S. 1331–1341, doi:10.2215/CJN.02740806.
- Bollee, Guillaume et al.: Presentation and Outcome of Patients with Systemic Amyloidosis Undergoing Dialysis. In: Clin J Am Soc Nephrol. Band 3, Nr. 2, 2008, S. 375–381, doi:10.2215/CJN.02470607.
- Berthold Jany, Tobias Welte: Pleuraerguss des Erwachsenen – Ursachen, Diagnostik und Therapie. In: Deutsches Ärzteblatt. Band 116, Nr. 21, (Mai) 2019, S. 377–385, hier: S. 379.
- L. M. Dember u. a.: Eprodisate for the Treatment of Renal Disease in AA Amyloidosis. In: N Engl J Med. Band 356, Nr. 23, 2007, S. 2349–2360, doi:10.1056/NEJMoa065644, PMID 17554116.
- A Phase II Open-label Randomized Study of Dietary Supplement With Epigallocatechin Gallate (EGCG) to Improve Cardiac Dysfunction in Patients With AL Amyloidosis Who do Not Require Chemotherapy (EpiCardiAL). ClinicalTrials.gov.
- S. O. Schönland: Fortschritte in der Diagnostik und Therapie der Amyloidosen. In: Deutsches Ärzteblatt. Band 103, Nr. 34–35, 2006, S. 2237 (aerzteblatt.de [PDF]).
- Dominotransplantation bei der Leber. (Nicht mehr online verfügbar.) nach Deutsche Stiftung für Organtransplantation, archiviert vom Original am 20. Oktober 2011; abgerufen am 5. August 2010.
- Derliz Mereles, Sebastian J. Buss, Stefan E. Hardt, Werner Hunstein, Hugo A. Katus: Effects of the main green tea polyphenol epigallocatechin-3-gallate on cardiac involvement in patients with AL amyloidosis. In: Clinical Research in Cardiology. Band 99, Nr. 8, 2010, S. 483–490, doi:10.1007/s00392-010-0142-x.
- Jan Bieschke, Jenny Russ, Ralf P. Friedrich, Dagmar E. Ehrnhoefer, Heike Wobst, Katja Neugebauer, Erich E. Wanker: EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. In: Proceedings of the National Academy of Sciences. Band 107, Nr. 17, 2010, S. 7710–7715, doi:10.1073/pnas.0910723107.
- FDA approves first-of-its kind targeted RNA-based therapy to treat a rare disease, PM FDA vom 10. August 2018, abgerufen am 21. April 2021
- Zusammenfassung der Merkmale des Arzneimittels. (PDF) EMA, abgerufen am 21. April 2021.
{kind=link}