Hyperparathyreoidismus
Hyperparathyreoidismus (HPT) ist eine Regulationsstörung der Epithelkörperchen (Nebenschilddrüsen). Der Hyperparathyreoidismus ist gekennzeichnet durch eine vermehrte Bildung von Nebenschilddrüsenhormon (Parathormon), welches den Calcium-Spiegel im Blut reguliert.
| Klassifikation nach ICD-10 | |
|---|---|
| E21.0 | Primärer Hyperparathyreoidismus |
| E21.1 | Sekundärer Hyperparathyreoidismus |
| E21.2 | Tertiärer, quartärer oder quintärer Hyperparathyreoidismus |
| ICD-10 online (WHO-Version 2019) | |
Liegt der vermehrten Bildung von Parathormon eine gutartige Geschwulst (Adenom) der Nebenschilddrüse zugrunde, spricht man von einer primären Überfunktion der Nebenschilddrüsen (primärer Hyperparathyreoidismus). Kennzeichen des primären Hyperparathyreoidismus sind ein erhöhter Parathormon-Spiegel und ein erhöhtes Serum-Calcium.
Ist die vermehrte Bildung von Parathormon die adäquate Reaktion der Nebenschilddrüsen auf ein vermindertes Serum-Calcium (Hypokalzämie, z. B. bei Vitamin-D-Mangel), spricht man von sekundärem Hyperparathyreoidismus. Charakteristisch für den sekundären Hyperparathyreoidismus ist ein erhöhter Parathormon-Spiegel bei niedrigem Serum-Calcium. Eine wichtige Ursache des sekundären Hyperparathyreoidismus ist die verminderte Aktivierung von Vitamin D aufgrund einer chronischen Nierenerkrankung.
Ein über lange Zeit bestehender sekundärer Hyperparathyreoidismus kann aufgrund einer chronischen Überstimulierung der Nebenschilddrüsen zu einem inadäquaten Anstieg des Parathormons führen. Es sind auch hier Parathormon-Spiegel und Serum-Calcium erhöht, aber man spricht von tertiärem Hyperparathyreoidismus. Vom primären Hyperparathyreoidismus kann der tertiäre Hyperparathyreoidismus durch die Krankenvorgeschichte abgegrenzt werden.
Folgen des Hyperparathyreoidismus, welche das Krankheitsbild der Ostitis fibrosa generalisata bilden, sind Abbau von Knochensubstanz aufgrund einer vermehrten Calcium-Freisetzung aus dem Knochen, Nierensteine aufgrund einer vermehrten Calcium-Ausscheidung in den Urin, Verkalkungen der Blutgefäße durch Ablagerung von Calcium und Phosphat sowie eine Vielzahl weiterer, zum Teil unspezifischer Symptome. Da eine Bestimmung des Serum-Calciums häufig im Rahmen einer Routine-Blutuntersuchung erfolgt, wird die Diagnose meist in einem frühen Stadium gestellt, in dem noch keine oder nur unspezifische Symptome bestehen.
Die Therapie des primären Hyperparathyreoidismus erfolgt durch operative Entfernung des Nebenschilddrüsen-Adenoms. Ist eine Operation nicht möglich oder wird diese nicht gewünscht, kann bei geringgradig erhöhtem Serum-Calcium der Krankheitsverlauf durch regelmäßige Kontrollen von Parathormon und Calcium beobachtet werden. Bei stark erhöhtem Calcium kann die Bildung von Parathormon durch das Medikament Cinacalcet gehemmt werden.
Der sekundäre Hyperparathyreoidismus wird behandelt mit Vitamin D, Cinacalcet und Phosphatbindern, letztere senken bei chronischer Nierenkrankheit erhöhte Phosphatspiegel.
Der tertiäre Hyperparathyreoidismus wird durch operative Entfernung der Nebenschilddrüsen behandelt. Um eine ausreichende Bildung von Parathormon zu gewährleisten, wird entweder ein Teil eines Epithelkörperchens belassen (subtotale Parathyreoidektomie) oder ein Teil eines Epithelkörperchens wird an anderer Stelle in einen Muskel eingepflanzt (autologe Retransplantation).
Die Nebenschilddrüsen (Epithelkörperchen)
Die Epithelkörperchen sind in etwa linsengroße Organe. Sie haben einen Durchmesser von 5 bis 8 mm und ein Gewicht von etwa 20 bis 50 mg. Sie liegen in der Regel jeweils hinten am oberen und unteren Pol der Schilddrüse. Selten können aber auch Nebenschilddrüsen im Bereich des Thorax gefunden werden. Der Grund findet sich in der embryologischen Entwicklung der Nebenschilddrüsen. Die meisten Menschen haben vier Epithelkörperchen. Sie bilden das Parathormon, ein Hormon, welches den Calciumspiegel im Körper reguliert.
Parathormon und Calcium-Haushalt
Parathormon und Calcitriol sind die beiden hauptsächlichen Hormone, welche den Calcium-Phosphat-Haushalt regulieren. Parathormon hält den Serum-Spiegel von Calcium in einem engen Bereich, kurzfristig durch vermehrte Rückresorption von Calcium im Tubulussystem der Niere und Förderung der Freisetzung von Calcium durch Abbau von Knochensubstanz. Längerfristig fördert Parathormon die Umwandlung von Calcidiol in Calcitriol und stimuliert so die Calcium-Aufnahme im Darm.
Die Sekretion von Parathormon wird durch den calciumsensitiven Rezeptor reguliert, welcher an der Oberfläche der Nebenschilddrüsen-Zellen exprimiert wird. Eine Zunahme der Konzentration an ionisiertem Calcium führt über eine Aktivierung des calciumsensitiven Rezeptors zu einer Hemmung der Parathormon-Sekretion, ein Abfall des ionisierten Calciums führt zu einer gesteigerten Sekretion von Parathormon.
Primärer Hyperparathyreoidismus
Pathogenese
Bei Patienten mit primärem Hyperparathyreoidismus ist die Parathormon-Sekretion in Relation zur Calcium-Konzentration im Serum inadäquat hoch, ausgelöst durch ein Adenom, Adenokarzinom oder eine Hyperplasie einer oder mehrerer Nebenschilddrüsen (Epithelkörperchen). Ursache der vermehrten Parathormon-Sekretion ist eine verminderte Sensitivität (Empfindlichkeit) des calciumsensitiven Rezeptors aufgrund einer verminderten Anzahl von calciumsensitiven Rezeptoren pro Nebenschilddrüsen-Zelle und/oder eine vermehrte Masse an Nebenschilddrüsen-Gewebe durch eine vermehrte Anzahl an Nebenschilddrüsen-Zellen. Die Regulation der Parathormon-Sekretion ist dabei nicht vollständig aufgehoben, es ist aber eine höhere Calcium-Konzentration erforderlich, um die Parathormon-Sekretion zu hemmen – der Soll-Wert der Calcium-Konzentration ist in höhere Bereiche verschoben.[1][2]
Ätiologie
Nur in seltenen Fällen kann eine Ursache des primären Hyperparathyreoidismus, ionisierende Strahlung oder genetische Veränderungen, gefunden werden.
Ionisierende Strahlung
Eine erhöhte Inzidenz von primärem Hyperparathyreoidismus fand sich nach Bestrahlung der Halsregion,[3] das Risiko, einen primären Hyperparathyreoidismus zu entwickeln steigt mit der Strahlendosis.[4] Auch bei Überlebenden des Atombombenabwurfs auf Hiroshima fand sich eine erhöhte Inzidenz an primärem Hyperparathyreoidismus.[5] Bei Patienten mit durch Strahlung ausgelöstem Hyperparathyreoidismus, ist die Inzidenz von Schilddrüsenkrebs erhöht, ansonsten unterscheidet sich der Verlauf aber nicht von Patienten mit idiopathischem Hyperparathyreoidismus (d. h. Hyperparathyreoidismus ohne nachweisbare Ursache).[6] Die Daten zum Auftreten eines primären Hyperparathyreoidismus nach Radiojodtherapie sind widersprüchlich, eine Studie fand ein gehäuftes Auftreten,[7] in einer weiteren Studie konnte ein Zusammenhang zwischen Radiojodtherapie und Hyperparathyreoidismus dagegen nicht bestätigt werden.[8]
Eine aktuelle Studie hat beobachtet, dass Liquidatoren, die im Jahre 1986 mit der Dekontamination des Kernreaktors in Tschernobyl beschäftigt waren, ein deutlich erhöhtes Risiko für die Entwicklung eines primären Hyperparathyreoidismus aufwiesen.[9]
Genetische Veränderungen
Bei primärem Hyperparathyreoidismus findet sich in der Regel eine monoklonale Vermehrung atypischer Nebenschilddrüsenzellen, d. h. die veränderten Zellen gehen auf eine einzige veränderte Mutterzelle zurück. Im veränderten Nebenschilddrüsengewebe wurden Mutationen in einer ganzen Reihe von Genen nachgewiesen: In Genen, welche das Zellwachstum kontrollieren, in Protoonkogenen sowie in Tumorsuppressorgenen.
Im Einzelnen wurden bisher Veränderungen in folgenden Genen beschrieben:
- CYCLIN D1 hat multiple Funktionen beim Zellwachstum, bei der Zelldifferenzierung und im Zellstoffwechsel. Eine erhöhte Aktivität von Cyclin D1 spielt eine wichtige Rolle bei der Entstehung von Tumoren einschließlich von Adenomen der Nebenschilddrüse.
- Multiple endokrine Neoplasie Typ I; MEN1: Inaktivierende Mutationen in MEN1, einem Tumorsuppressorgen führen zu autosomal-dominant vererbten Neoplasien von Nebenschilddrüsen, der Hypophyse und der Inselzellen im Pankreas sowie zu vermehrtem Auftreten von Magengeschwüren.
- HRPT2 codiert für das Protein Parafibromin, das im Komplex mit weiteren Proteinen (PAF1 und CTR9) an RNA-Polymerase II und an Histon-Methyltransferase bindet. Mutationen in HRPT2 führen zu Adenomen der Nebenschilddrüsen in Verbindung mit Kiefer-Tumoren, sowie zu Nebenschilddrüsenkarzinom.
- RET codiert für eine Rezeptor-Tyrosinkinase. Mutationen führen unter anderem zur multiplen endokrinen Neoplasie Typ IIA. Die betroffenen Patienten entwickeln C-Zell-Karzinome der Schilddrüse, Phäochromozytome und in 15 bis 20 % der Patienten einen primären Hyperparathyreoidismus.
Klinik
Der erhöhte Parathormonspiegel führt zu einem gesteigerten Knochenabbau und damit zu einer erhöhten Calcium-Konzentration im Blut. Der Knochen wird demineralisiert. Knochenschmerzen können auftreten. In der Niere sorgt das Parathormon für eine verminderte Calciumausscheidung mit dem Urin, so dass die Calciummenge im Blut zusätzlich ansteigt. Durch die Rückresorption von Calcium aus dem Urin kann das Löslichkeitsprodukt überschritten werden. In der Folge entstehen Nierensteine. Außerdem können Gallensteine und Entzündungen der Bauchspeicheldrüse auftreten. Die Beschwerden des Hyperparathyreoidismus werden mit den drei Worten „Stein-, Bein- und Magenpein“ umschrieben.
Sekundärer Hyperparathyreoidismus
Ursache hier ist eine verstärkte Hormonproduktion als Reaktion auf einen erhöhten Calciumverlust (Calciummangel, Hypokalzämie) des Körpers. Durch die kompensatorisch vermehrte Hormonproduktion kann die Calcium-Konzentration im Blut im unteren Normalbereich liegen.
Zugrunde liegende Erkrankungen des sekundären Hyperparathyreoidismus:
- Chronische Niereninsuffizienz (typisch: Calciumspiegel niedrig, Phosphatspiegel zu hoch)
- Malassimilationssyndrom: Störung der Calcium-Aufnahme im Darm.
- Leberzirrhose: Gestörte Umwandlung von Vitamin D3 (Cholecalciferol) in 25-Hydroxycholecalciferol in der Leber. Durch das fehlende aktive Vitamin D3 wird im Darm weniger Calcium aufgenommen und in der Niere weniger Calcium reabsorbiert.
- Cholestase: Durch den Mangel an Gallensäuren ist die Resorption von Vitamin D3 aus der Nahrung gestört.
- Fehlende Sonnenlichtexposition: In der Haut kann Vitamin D3 nicht mehr aus Cholesterin (bzw. 7-Dehydro-Cholesterin) gebildet werden.
- Rachitis
Tertiärer Hyperparathyreoidismus
Wenn über einen längeren Zeitraum ein sekundärer Hyperparathyreoidismus besteht, wird außer der Funktion der Nebenschilddrüsen auch deren Wachstum stimuliert, so dass es schließlich zu einer autonomen Überproduktion in den Epithelkörperchen kommen kann. Wie beim primären Hyperparathyreoidismus ist der Regelkreis an der Stelle der Nebenschilddrüsen aufgetrennt.
Quartärer und quintärer Hyperparathyreoidismus
Selten sind quartäre und quintäre Formen eines Hyperparathyreoidismus. Als quartärer Hyperparathyreoidismus wird ein sekundärer Hyperparathyreoidismus auf dem Boden einer Nierenschädigung bezeichnet, wenn die Nierenschädigung ihrerseits durch einen primären Hyperparathyreoidismus verursacht worden war. Pathophysiologisch gesehen ist der Regelkreis wie beim sekundären Hyperparathyreoidismus an der Niere aufgetrennt. Als quintär wird ein Hyperparathyreoidismus bezeichnet, wenn die Entkoppelung der Parathormonsekretion aus einem oder mehreren verbliebenen Epithelkörperchen auf einem langjährigen quartären Hyperparathyreoidismus beruht. Differentialdiagnostisch muss in beiden Situationen die Möglichkeit eines schlafenden Nebenschilddrüsenadenoms bedacht werden.[10]
Symptome des Hyperparathyreoidismus
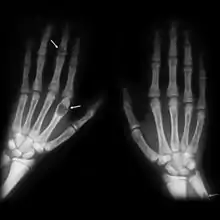
Bei Diagnosestellung eines primären Hyperparathyreoidismus können folgende Krankheitsmanifestationen bestehen:
- zufällig entdeckte Erhöhung des Calciumspiegels im Blut, ohne dass klinische Krankheitszeichen bestehen (asymptomatische Hyperkalziämie),
- Erhöhung des Calciumspiegels im Blut mit klinischen Krankheitszeichen (symptomatische Hyperkalziämie),
- Folgekrankheiten des Hyperparathyreoidismus: verminderte Knochendichte (Osteopenie), Knochenschwund (Osteoporose), Nierensteine (Nephrolithiasis),
- selten typische Knochenveränderungen (Ostitis fibrosa cystica) oder hyperparathyreotische Krise.
In früheren Jahren standen bei Erstmanifestation meist die klassischen Symptome der fortgeschrittenen Erkrankung im Vordergrund: Stein-Bein-Magen-Pein durch Nierensteine, pathologische Frakturen und Magengeschwüre. Seit der Zunahme von Blutuntersuchungen und Knochendichtemessungen überwiegen symptomlose (asymptomatische) und symptomarme (oligosymptomatische) Verläufe.[11]
Für die generalisierte Demineralisation mit braunen Tumoren im fortgeschrittenen Stadium der Erkrankung war früher die Bezeichnung Engel-von Recklinghausen Syndrom[12] oder Recklinghausensche Krankheit gebräuchlich.
Asymptomatischer Hyperparathyreoidismus
Mindestens 80 % der Patienten mit primärem Hyperparathyreoidismus werden durch den Zufallsbefund eines erhöhten Serum-Calciums diagnostiziert. Diese Patienten haben meist eine milde (im Mittel 2,8 mmol/l, Norm 2,1–2,6 mmol/l) und intermittierende Hyperkalziämie.[13] Gelegentlich sind die Calcium-Spiegel normal, und der primäre Hyperparathyreoidismus wird aufgrund einer verminderten Knochendichte festgestellt.[11]Bei genauerer Anamnese geben manche Patienten unspezifische Symptome an wie Appetitlosigkeit (Anorexie), milde depressive Verstimmung,[14] milde Gedächtnis- oder neuromuskuläre Störungen.[15]
Häufig besteht bei primärem Hyperparathyreoidismus ein Vitamin-D-Mangel, der die Diagnose verschleiern kann. Nach Gabe von Vitamin D kann es dann zu einem Anstieg des Calciumspiegels kommen, der auf die Diagnose hinweist.[16] Bei Vitamin-D-Mangel und Hyperparathyreoidismus kann es aber auch zu ausgeprägteren Krankheitsbildern kommen mit größeren Nebenschilddrüsen-Adenomen, höheren Konzentrationen an Parathormon, erhöhtem Knochenumbau und vermehrten Knochenbrüchen.[17]
Symptomatischer Hyperparathyreoidismus
Durch den erhöhten Parathormonspiegel wird Calcium aus den Knochen abgebaut. Die Knochendichte nimmt ab, Calciumspiegel im Blut und Calciumausscheidung im Urin steigen an.
Der erhöhte Calciumspiegel im Blut (Hyperkalzämie) führt zu unspezifischen Beschwerden Appetitlosigkeit (Anorexie), Übelkeit (Nausea), Verstopfung (Obstipation), gesteigerter Durst (Polydipsie) und vermehrte Harnproduktion (Polyurie).
Am Knochen kommt es zu Osteoporose und pathologischen Frakturen, insbesondere im Bereich der Wirbelsäule.[18]
Bei langjährigem sekundärem renalen Hyperparathyreoidismus können in den Knochen sog. braune Tumoren auftreten, die aus knochen-abbauenden Zellen (Osteoklasten) und Bindegewebe bestehen. Die Bezeichnung brauner Tumor bezieht sich auf die braune Färbung, die von Einblutungen hervorgerufen wird.[19]
In den Nieren führt die erhöhte Calciumausscheidung zum Ausfällen von Nierensteinen (Nephrolithiais), Weichteilverkalkungen (Nephrokalzinose), Nierenkoliken, chronischem Nierenversagen und Störungen der Tubulusfunktion. Die Nierensteine bestehen vorwiegend aus Calciumoxalat, seltener Calciumphosphat. Bei etwa 15–20 % der Patienten mit primärem Hyperparathyreoidismus liegen Nierensteine vor, dem gegenüber findet sich bei etwa 5 % der Nierensteinträger ein primärer Hyperparathyreoidismus als Ursache des Steinleidens.[20]
Häufig klagen Patienten über neuromuskuläre Beschwerden wie Schwäche, Abgeschlagenheit und Müdigkeit.[21]
Zudem treten bei Patienten mit primärem Hyperparathyreoidismus häufiger als in der Normalbevölkerung psychiatrische Erkrankungen auf wie Teilnahmslosigkeit (Lethargie), Depressionen, Verlust des Realitätsbezuges (Psychosen), verminderte soziale Interaktionsfähigkeit und Einschränkung der kognitiven Fähigkeiten (Demenz). Exakte Zahlen hierzu liegen aber nicht vor.[22]
Es besteht eine erhöhte Prävalenz an Bluthochdruck,[23] Übergewicht[24] und gestörter Glukosetoleranz[25] sowie an rheumatologischen Erkrankungen wie erhöhter Harnsäure (Hyperurikämie), Gicht und Pseudogicht.[26] Es gibt Hinweise darauf, dass die Prävalenz von Krebserkrankungen bei primärem Hyperparathyreoidismus erhöht ist, und das erhöhte Krebsrisiko auch nach operativer Entfernung des Nebenschilddrüsenadenoms fortbesteht. Aufgrund dieser Befunde wird über gemeinsame prädisponierende Faktoren für Hyperparathyreoidismus und Krebserkrankungen spekuliert.[27][28][29]
Bei Patienten mit primärem Hyperparathyreoidismus besteht eine hohe Inzidenz an kardiovaskuläre Erkrankungen, insbesondere linksventrikuläre Hypertrophie sowie Verkalkungen von Herzmuskel (Myokard), Aorten- und Mitralklappe. Das Risiko, an einer Herzerkrankung zu versterben, ist möglicherweise erhöht.[30][31]
Bereits bei mildem primärem Hyperparathyreoidismus kommt es zu einer Versteifung der Blutgefäße.[32][33]
Insbesondere beim sekundären renalen Hyperparathyreoidismus kann es zu schweren Verkalkungen der Gefäße kommen, darunter auch der Herzkranzgefäße. Die Folgen sind eine erheblich erhöhte Morbidität und Mortalität an kardiovaskuläre Erkrankungen.[34][35]
Die enge Verzahnung zwischen Knochen- und Herzkreislauferkrankung beim sekundären Hyperparathyreoidismushat hat dazu geführt, dass in den neuesten Leitlinien der Begriff Renale Osteopathie durch den Begriff Renale Mineral- und Knochenerkrankung (Chronic kidney disease-mineral and bone disorder) ersetzt wurde, welcher sowohl Knochenstoffwechselstörung als auch Gefäßverkalkungen und deren Folgen beinhaltet.[36]
Eine besonders schwere Komplikation des sekundären Hyperparathyreoidismus ist die (Calciphylaxie), bei der Calciumablagerungen zu Thrombosen in den Arteriolen der Haut führen. Diese können schwere Durchblutungsstörungen mit ausgedehnten Haut-Nekrosen hervorrufen, die häufig zum Tode führen.[37]
Außerdem wird die Gastrinproduktion angeregt, so dass es zu einer Magenschleimhautentzündung (Gastritis) und Zwölffingerdarmgeschwür (Ulcus duodeni) kommen kann.[38]
Epidemiologie
Die Häufigkeit des primären Hyperparathyreoidismus hat in den letzten Jahren abgenommen. In einem amerikanischen Register betrug die Inzidenz im Zeitraum von 1993 bis 2001 21,6 Fälle pro 100,000 Personen-Jahre, im Zeitraum von 1983 bis 1992 29,1 Fälle und von 1974 bis 1982 82,5 Fälle. Die Gründe für diese Abnahme der Häufigkeit sind nicht bekannt, mögliche Ursachen sind eine verminderte Exposition gegenüber ionisierender Strahlung und eine verbesserte Versorgung mit Vitamin D.
Der primäre Hyperparathyreoidismus kann in jedem Lebensalter auftreten, die meisten Erkrankungen manifestieren sich aber erst nach dem 45. Lebensjahr. Frauen sind etwa doppelt so oft betroffen wie Männer, möglicherweise weil der nach der Menopause eintretende Knochenabbau einen latenten Hyperparathyreoidismus demaskieren kann.[39]
Histologie
Nebenschilddrüse
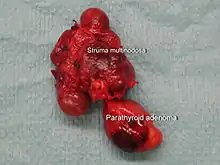
Bei primärem Hyperparathyreoidismus werden bei der feingeweblichen Untersuchung der Nebenschilddrüsen folgende Veränderungen beschrieben:[40]
- In 89 % der Fälle von primärem Hyperparathyreoidismus wird eine einzelne gutartige Geschwulst (Adenom) der Nebenschilddrüse gefunden, in etwa 5 % der Fälle finden sich zwei Adenome.[41] Die meisten Adenome werden von den Hauptzellen der Nebenschilddrüse gebildet und sind von einer Kapsel umgeben. Gelegentlich werden Adenome gefunden, die aus oxyphilen Zellen bestehen, diese Adenome sind in der Regel größer. Es gibt auch Parathormon-bildende Adenome im Thymus.[42]
- In 6 % der Fälle findet sich eine glanduläre Hyperplasie, d. h. eine diffuse Vergrößerung aller vier Nebenschilddrüsenkörperchen durch Vermehrung der Hauptzellen. Sehr selten wird eine glanduläre Hyperplasie durch eine Vermehrung von Klarzellen hervorgerufen.[43]
- In ca. 2 % der Fälle findet sich ein Nebenschilddrüsenkarzinom, das gekennzeichnet ist durch Invasion von Kapsel und Gefäßen, Lymphknoten- und Fernmetastasen.[44]
Knochen
Bei der feingeweblichen Untersuchung des Knochens finden sich Veränderungen, die als Ostitis fibrosa beschrieben werden. Der erhöhte Parathormon-Spiegel aktiviert Osteoklasten. Die vermehrte Osteoklasten-Tätigkeit führt zu einem Abbau von Knochensubstanz. Die feingewebliche Untersuchung zeigt Mikrofrakturen und Einblutungen. Es bilden sich Hohlräume, die gefüllt sind mit Bindegewebe, Osteoklasten und Hämosiderin-beladenen Makrophagen. Zunehmende Auflösung (Resorption) von Knochengewebe und bindegewebiger Umbau (Fibrose) führt zur Bildung von Knochenzysten, die mit bloßem Auge sichtbar sind. Bei weiterem Fortschreiten der Erkrankung verschmelzen die Knochenzysten zu braunen Tumoren, die braune Färbung ist Folge von Einblutungen und Hämosiderinablagerungen (Fallbeispiele und Abb. unter[45][46])
Diagnose
Diagnose des primären Hyperparathyreoidismus
Erster Hinweis auf das Vorliegen eines primären Hyperparathyreoidismus ist ein erhöhter Calcium-Spiegel (Hyperkalzämie). Andere Ursachen einer Hyperkalzämie können durch Bestimmung der Parathormon-Konzentration abgegrenzt werden.[13] Die Diagnose eines primären Hyperparathyreoidismus wird gestellt, wenn das Parathormon erhöht ist, oder wenn das Parathormon zwar im Normalbereich von 10 bis 60 pg/ml liegt, aber relativ erhöht ist, bezogen auf einen gleichzeitig erhöhten Calcium-Spiegel.[47] Etwa 80–90 % der Patienten mit primärem Hyperparathyreoidismus haben ein erhöhtes Parathormon, bei 10–20 % der Patienten liegt das Parathormon im Normalbereich.[48]
Bei primärem Hyperparathyreoidismus werden osteolytische Veränderungen im Mund-, Kiefer- und Gesichtsbereich im Gegensatz zum übrigen Skelettsystem, z. B. Akroosteolysen, selten beobachtet, können aber den ersten klinischen Hinweis auf die endokrine Erkrankung darstellen.[49]
Differentialdiagnose des primären Hyperparathyreoidismus
Häufigste Ursache einer Hyperkalzämie sind neben dem Hyperparathyreoidismus bösartige Krebserkrankungen. Meist ist bei Auftreten einer Hyperkalzämie die Krebserkrankung bekannt und weit fortgeschritten, zudem sind die Calciumspiegel bei Krebserkrankungen höher und die damit verbundenen Symptome schwerer. Das Parathormon ist bei Hyperkalzämie aufgrund einer Krebserkrankung meist sehr niedrig.
Andere Ursachen einer Hyperkalzämie wie Milch-Alkali-Syndrom, Sarkoidose und Vitamin-D-Überdosierung können ebenfalls durch ein erniedrigtes Parathormon vom Hyperparathyreoidismus abgegrenzt werden.
Bei der familiären hypokalzurischen Hyperkalzämie (FHH) ist die Calcium-Ausscheidung im Urin vermindert, bei Patienten mit primärem Hyperparathyreoidismus ist die Calcium-Ausscheidung dagegen normal oder erhöht.[50]
Thiaziddiuretika vermindern die Calciumausscheidung über die Nieren und führen zu einer milden Hyperkalzämie. Bei Patienten mit mildem Hyperparathyreoidismus können Thiaziddiuretika die Grunderkrankung demaskieren, in dem sie zu einem deutlichen Calcium-Anstieg im Serum führen. Fällt ein erhöhter Calcium-Spiegel nach Absetzen von Thiaziddiuretika nicht ab, spricht dies für das Vorliegen eines primären Hyperparathyreoidismus.
Lithium verstärkt die Parathormon-Sekretion und vermindert die Calciumausscheidung über die Nieren, es kommt zu Hyperkalzämie und Hypokalzurie, einige Patienten weisen erhöhte Parathormon-Spiegel auf. Nach Absetzen des Lithium kann sich die Hyperkalzämie normalisieren, nach längerer (über 10 Jahre) dauernder Behandlung mit Lithium ist eine Normalisierung der Calcium-Spiegel nach Beendigung der Therapie weniger wahrscheinlich.
Diagnose des sekundären Hyperparathyreoidismus
Der sekundäre Hyperparathyreoidismus ist charakterisiert durch einen erniedrigten Calcium-Spiegel mit adäquatem Anstieg des Parathormons.
Häufigste Ursache des sekundären Hyperparathyreoidismus ist eine verminderte Produktion von aktiviertem Vitamin D (Calcitriol) aufgrund einer chronischen Nierenfunktionseinschränkung.
Weitere Ursachen eines sekundären Hyperparathyreoidismus sind:
- verminderte Calcium-Zufuhr mit der Nahrung
- verminderte Calcium-Aufnahme über den Darm (Calcium-Malabsorption)
- erhöhtes Phosphat (Hyperphosphatämie)
- Vitamin-D-Mangel
- Adipositaschirurgie
- Zöliakie
- Exokrine Pankreasinsuffizienz
- Calciumverluste über die Nieren
- Idiopathische Hyperkalzurie
- Schleifendiuretika
- Verminderter Knochenumbau, z. B. aufgrund von Bisphosphonaten
- Hungry bone syndrome – Syndrom des „hungrigen Knochens“
Differentialdiagnose des sekundären Hyperparathyreoidismus
Einige Patienten mit primärem Hyperparathyreoidismus haben normales Serum-Calcium (Normocalzämischer primärer Hyperparathyreoidismus). In diesen Fällen ist die Abgrenzung zwischen primärem und sekundärem Hyperparathyreoidismus schwierig. Häufig ist eine verminderte Knochendichte erste Manifestation der Erkrankung.
Ursachen des normocalzämischen primären Hyperparathyreoidismus sind:
- Frühstadium oder unvollständige Ausprägung („forme fruste“) der Erkrankung; im Verlauf von 3 Jahren entwickeln etwa 40 % der Betroffenen weitere Krankheits-Manifestationen (in der Hälfte der Fälle Hyperkalzämie, ansonsten Nierensteine, Knochenbrüche, Hyperkalzurie, Osteoporose).[51] Verlaufskontrollen sind daher angezeigt.
- Primärer Hyperparathyreoidismus bei gleichzeitigem Vitamin-D-Mangel
Klinische Chemie
Die Diagnose wird durch Laboruntersuchungen gestellt.
- Ein erhöhtes oder im hohen Normalbereich liegendes Parathormon weist bei gleichzeitig erhöhtem Serum-Calcium auf einen primären Hyperparathyreoidismus hin.
- Die Calcium-Ausscheidung im 24 Stunden Sammelurin ist bei ca. 40 % der Patienten mit primärem Hyperparathyreoidismus erhöht, bei ca. 60 % normal. Bei einer Calcium-Ausscheidung über 400 mg/24 h steigt das Risiko von Nierensteinen. Eine verminderte Calcium-Ausscheidung findet sich bei Familiärer hypokalzurischer Hyperkalzämie oder bei primärem Hyperparathyreoidismus bei gleichzeitig bestehendem mit Vitamin-D-Mangel.
- Durch einen verminderten Blutspiegel an 25(OH)Vitamin D3 kann ein primärer Hyperparathyreoidismus bei gleichzeitig bestehendem mit Vitamin-D-Mangel von einer Familiären hypokalzurischen Hyperkalzämie abgegrenzt werden.
- Bei primärem Hyperparathyreoidismus liegen die Serumspiegel von Calcitriol im hochnormalen Bereich oder sind erhöht, bei sekundärem Hyperparathyreoidismus aufgrund eines chronischen Nierenversagens sind die Calcitriol-Spiegel vermindert.
- Bei primärem Hyperparathyreoidismus ist die Phosphat-Ausscheidung über die Nieren erhöht, der Phosphatspiegel im Serum ist vermindert (Hypophosphatämie) oder liegt im niedrig normalen Bereich. Bei sekundärem renalem Hyperparathyreoidismus ist der Phosphatspiegel typischerweise erhöht.
- Ein erhöhtes Serum-Kreatinin weist auf eine Nierenfunktionseinschränkung. Diese kann Folge der Hyperkalzämie bei primärem Hyperparathyreoidismus sein aber auch Ursache eines sekundären Hyperparathyreoidismus.
- Biochemisch Marker des Knochenumsatzes (z. B. die Alkalische Phosphatase) liegen bei primärem und sekundärem Hyperparathyreoidismus im hochnormalen oder erhöhten Bereich.
Osteodensitometrie
Bei Patienten mit primärem Hyperparathyreoidismus ist der Mineralsalzgehalt des Knochens vermindert. Die Knochendichtemessung (Osteodensitometrie) ist nicht zur Diagnosestellung erforderlich, sondern zur Beurteilung der Schwere der Erkrankung, zur Verlaufsbeobachtung und zur Entscheidung, ob eine konservative oder eine operative Therapie angezeigt ist.
Bei Patienten mit sekundärem renalem Hyperparathyreoidismus besteht keine enge Beziehung zwischen Knochenbrüchen und Knochendichte. Eine routinemäßige Knochendichtemessung ist bei renalem Hyperparathyreoidismus nach dem derzeitigen Kenntnisstand nicht sinnvoll.[52][53]
Lokalisationsdiagnostik
Vor einer geplanten Operation können Lokalisation und Größe der betroffenen Nebenschilddrüsenkörperchen durch Sonografie, Szintigrafie mit Technetium-99m-Sestamibi (Nebenschilddrüsenszintigrafie), Computertomographie oder Kernspintomografie dargestellt werden.
Therapie des Hyperparathyreoidismus
Chirurgische Therapie
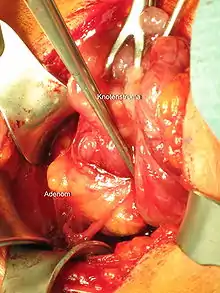
Die chirurgische Entfernung des (oder aller) vergrößerten Epithelkörperchens, führt im Vergleich zur Verlaufsbeobachtung zu einer Verbesserung von Knochendichte und Lebensqualität[54] sowie zu einer Verminderung von Knochenbrüchen, insbesondere von Schenkelhalsfrakturen.[55] In der Mehrzahl der asymptomatischen Patienten finden sich aber auch bei langer Verlaufsbeobachtung keine Hinweise auf ein Fortschreiten der Erkrankung wie Abnahme von Knochendichte, Anstieg von Calcium und Parathormon oder Auftreten von Nierensteinen.
Aus diesem Grund wurden Leitlinien entwickelt, deren Ziel ist, diejenigen Patienten einer chirurgischen Therapie zuzuführen, bei denen das Risiko für Organschäden oder ein Fortschreiten der Erkrankung am höchsten ist.
Kriterien des 2002 NIH Workshop on Asymptomatic Primary Hyperparathyroidism[56] zur chirurgischen Therapie:
- Serum Calcium 0,25 mmol/l (1,0 mg/dl) über der oberen Normgrenze.
- Calcium Ausscheidung im Urin über 10 mmol/Tag (400 mg/Tag) bei normaler Kost.
- Kreatinin-Clearance 30 % des Altersdurchschnitts oder weniger.
- Knochendichte an Hüfte, Lendenwirbelsäule oder Radius mehr als 2,5 Standardabweichungen unter dem Mittel (T-Wert < −2,5).
- Alter unter 50 Jahren.
- Regelmäßige Verlaufskontrollen nicht gewährleistet.
Nicht chirurgische Therapie
Obwohl die chirurgische Therapie zu einer definitiven Heilung des primären Hyperparathyreoidismus führt, werden nicht alle Patienten operiert, insbesondere bei Fehlen von Symptomen, nur geringer Erhöhung des Calciums, bei einem Lebensalter über 50 Jahren, wenn schwere Begleiterkrankungen bestehen oder wenn eine Operation nicht gewünscht wird. In diesen Fällen stehen folgende nicht operative Verfahren zur Verfügung:
- Bisphosphonate hemmen den Knochenabbau durch Osteoklasten. Alendronat führt bei asymptomatischem primären Hyperparathyreoidismus zu einer Zunahme der Knochenmasse. Risedronat senkt erhöhte Calcium-Spiegel.
- Raloxifen ist ein selektiver Östrogenrezeptor-Modulator. Bei postmenopausalen Frauen mit primärem Hyperparathyreoidismus senkt Raloxifen das Serum-Calcium. Nach sorgfältiger Nutzen-Risiko-Abwägung kann alternativ eine Östrogen-Ersatztherapie in Erwägung gezogen werden.
- Seit 2004 sind Calcimimetika (z. B. Cinacalcet) verfügbar, die am calciumsensitiven Rezeptor der Nebenschilddrüsenzelle angreifen und somit direkt die Parathormonausscheidung hemmen.
- Perkutane Alkoholablation: Durch Injektion von Alkohol in das vergrößerte Nebenschilddrüsenkörperchen kann dieses zerstört werden. Nebenwirkungen sind eine Schädigung des Nervus laryngeus recurrens und ein zu starkes Absinken des Serum-Calciums (Hypokalzämie)
- Darüber hinaus werden eine calciumarme Ernährung mit ausreichendem Gehalt an Vitamin D sowie regelmäßige körperliche Betätigung empfohlen[57]
Bei Patienten, bei denen keine Operation durchgeführt wird, werden Calcium halbjährlich, Kreatinin und Knochendichte jährlich kontrolliert. Bei Hinweisen auf ein Fortschreiten der Erkrankung wird operiert.
Therapie des sekundären Hyperparathyreoidismus
Der sekundäre Hyperparathyreoidismus wird im Allgemeinen durch Beseitigung der Grunderkrankung behandelt. Für den sekundäre Hyperparathyreoidismus infolge eines chronischen Nierenversagens existiert eine abgestufte, durch Leitlinien festgelegte medikamentöse Therapie.
Sekundärer Hyperparathyreoidismus bei chronischem Nierenversagen
Der sekundäre Hyperparathyreoidismus bei chronischem Nierenversagen wird mit phosphatarmer Diät, Vitamin-D-Metaboliten, Phosphatbindern und Cinacalcet behandelt.[58][59]
Liegt der Parathormonspiegel im Stadium 2–3 des chronischen Nierenversagens über 70 pg/ml (bzw. > 110 pg/ml im Stadium 4 oder > 300 pg/ml im Stadium 5) wird mit einer phosphatreduzierten Ernährung begonnen. Gelingt es nicht, durch eine phosphatarme Diät den Phosphatspiegel ausreichend zu senken, werden Medikamente eingesetzt, welche das mit der Nahrung zugeführte Phosphat im Darm binden (orale Phosphatbinder): Calciumcarbonat, Calciumacetat, Aluminiumhydroxid, Sevelamer oder Lanthancarbonat. Calciumhaltige Phosphatbinder werden wegen der Gefahr der Weichteilverkalkung nicht bei erhöhten Calcium-Werten eingesetzt, die Menge ist begrenzt auf 2,5 g Calcium pro Tag. Aluminiumhaltige Phosphatbinder werden wegen der Gefahr der Aluminiumablagerung in Knochen und Gehirn nur bei sehr hohen Phosphatspiegeln und über einen begrenzten Zeitraum angewandt.
Ein verminderter Vitamin-D-Spiegel im Serum, (25(OH)Vitamin-D3 < 30 ng/ml) wird durch Gabe von Vitamin D ausgeglichen. Sinkt der Parathormonspiegel trotz dieser Maßnahmen nicht in den Zielbereich, wird zusätzlich aktives Vitamin D (Alfacalcidol oder Calcitriol) verabreicht. Nebenwirkung der Therapie mit aktivem Vitamin D ist ein Anstieg des Calcium- und Phosphatspiegels mit der Gefahr von Weichteilverkalkungen. Diese Nebenwirkung ist geringer bei Verwendung von pharmakologisch veränderten Vitamin D Metaboliten wie Paricalcitol.
Führen phosphatarme Diät, Phosphatbinder und Vitamin-D-Metaboliten nicht zum Erfolg, wird Cinacalcet, ein Antagonist des calciumsensitiven Rezeptors eingesetzt.
Ohne Therapie ist ein Übergang in einen tertiären Hyperparathyreoidismus wahrscheinlich.
Therapie des tertiären Hyperparathyreoidismus
Steigt der Parathormonspiegel trotz medikamentöser Therapie auf Werte über 1000 pg/ml und kommt es zu Knochenveränderungen und therapierefraktärer Hyperkalzämie und/oder Hyperphosphatämie müssen alle 4 (bis 8) Epithelkörperchen entfernt (Parathyroidektomie) und anschließend ein Teil eines Epithelkörperchens in Unterarm oder Musculus sternocleidomastoideus autotransplantiert werden, da sonst gar keine Parathormonbildung mehr stattfinden könnte. Alternativ kann auch eine subtotale Parathyroidektomie erfolgen, d. h. ein Nebenschilddrüsenkörperchen wird nicht vollständig entfernt.[60]
Siehe auch
Leitlinien
- John P. Bilezikian u. a.: Summary Statement from a Workshop on Asymptomatic Primary Hyperparathyroidism: A Perspective for the 21st Century. In: J Clin Endocrinol Metab. Nr. 12, 2002, S. 5353–5361 (Artikel).
- KDOQI Clinical Practice Guidelines for Bone Metabolism and Disease in Chronic Kidney Disease (Memento vom 14. Februar 2015 im Internet Archive).
Literatur
- Ludwig Weissbecker: Hyperparathyreoidismus (Ostitis fibrosa generalisata, Recklinghausensche Krankheit). In: Ludwig Heilmeyer (Hrsg.): Lehrbuch der Inneren Medizin. Springer-Verlag, Berlin/Göttingen/Heidelberg 1955; 2. Auflage ebenda 1961, S. 1057–1060.
Weblinks
- Hyperparathyreoidismus Pathologie – Bilddatenbank Pathopic der Universität Basel (PathoPic-Anleitung (PDF; 2,2 MB))
Einzelnachweise
- E. M. Brown: Four-parameter model of the sigmoidal relationship between parathyroid hormone release and extracellular calcium concentration in normal and abnormal parathyroid tissue. In: The Journal of Clinical Endocrinology and Metabolism. Band 56, Nr. 3, März 1983, ISSN 0021-972X, S. 572–581, PMID 6822654.
- J. H. Brossard, S. Whittom, R. Lepage, P. D’Amour: Carboxyl-terminal fragments of parathyroid hormone are not secreted preferentially in primary hyperparathyroidism as they are in other hypercalcemic conditions. In: The Journal of Clinical Endocrinology and Metabolism. Band 77, Nr. 2, August 1993, ISSN 0021-972X, S. 413–419, PMID 8345045.
- L. E. Tisell, S. Carlsson, M. Fjälling, G. Hansson, S. Lindberg, L. M. Lundberg, A. Odén: Hyperparathyroidism subsequent to neck irradiation. Risk factors. In: Cancer. Band 56, Nr. 7, 1. Oktober 1985, ISSN 0008-543X, S. 1529–1533, PMID 4027889.
- A. B. Schneider, T. C. Gierlowski, E. Shore-Freedman, M. Stovall, E. Ron, J. Lubin: Dose-response relationships for radiation-induced hyperparathyroidism. In: The Journal of Clinical Endocrinology and Metabolism. Band 80, Nr. 1, Januar 1995, ISSN 0021-972X, S. 254–257, PMID 7829622.
- S. Fujiwara, R. Sposto, H. Ezaki, S. Akiba, K. Neriishi, K. Kodama, Y. Hosoda, K. Shimaoka: Hyperparathyroidism among atomic bomb survivors in Hiroshima. In: Radiation Research. Band 130, Nr. 3, Juni 1992, ISSN 0033-7587, S. 372–378, PMID 1594765.
- S. Tezelman, J. M. Rodriguez, W. Shen, A. E. Siperstein, Q. Y. Duh, O. H. Clark: Primary hyperparathyroidism in patients who have received radiation therapy and in patients who have not received radiation therapy. In: Journal of the American College of Surgeons. Band 180, Nr. 1, Januar 1995, ISSN 1072-7515, S. 81–87, PMID 8000660.
- Shanthi M Colaço, Ming Si, Emily Reiff, Orlo H Clark: Hyperparathyroidism after radioactive iodine therapy. In: American Journal of Surgery. Band 194, Nr. 3, September 2007, ISSN 0002-9610, S. 323–327, doi:10.1016/j.amjsurg.2007.04.005, PMID 17693276.
- M. Fjälling, A. Dackenberg, I. Hedman, L. E. Tisell: An evaluation of the risk of developing hyperparathyroidism after 131I treatment for thyrotoxicosis. In: Acta Chirurgica Scandinavica. Band 149, Nr. 7, 1983, ISSN 0001-5482, S. 681–686, PMID 6650083.
- Bernhard O. Boehm, Silke Rosinger, David Belyi, Johannes W. Dietrich: The Parathyroid as a Target for Radiation Damage. In: New England Journal of Medicine. Band 365, Nr. 7, August 2011, S. 676–678, doi:10.1056/NEJMc1104982, PMID 21848480.
- S. Yavuz, W. F. Simonds, L. S. Weinstein, M. T. Collins, E. Kebebew, N. Nilubol, G. Q. Phan, S. K. Libutti, A. T. Remaley, M. Van Deventer, S. J. Marx: Sleeping parathyroid tumor: rapid hyperfunction after removal of the dominant tumor. In: J Clin Endocrinol Metab. 97(6), Jun 2012, S. 1834–1841, doi:10.1210/jc.2011-3030, Epub 16. April 2012. PMID 22508712
- John P Bilezikian, John T Potts: Asymptomatic primary hyperparathyroidism: new issues and new questions-bridging the past with the future. In: Journal of Bone and Mineral Research. 17 Suppl 2, November 2002, ISSN 0884-0431, S. N57–N67, PMID 12412779.
- Who named it
- S. J. Silverberg, J. P. Bilezikian: Evaluation and management of primary hyperparathyroidism. In: The Journal of Clinical Endocrinology and Metabolism. Band 81, Nr. 6, Juni 1996, ISSN 0021-972X, S. 2036–2040, PMID 8964825.
- Yuko Chiba, Katsuhiko Satoh, Satoshi Ueda, Nobuo Kanazawa, Yoshiaki Tamura, Toshiyuki Horiuchi: Marked improvement of psychiatric symptoms after parathyroidectomy in elderly primary hyperparathyroidism. In: Endocrine Journal. Band 54, Nr. 3, Juni 2007, ISSN 0918-8959, S. 379–383, doi:10.1507/endocrj.K06-152, PMID 17420608.
- E. Lundgren, E. Szabo, S. Ljunghall, R. Bergström, L. Holmberg, J. Rastad: Population based case-control study of sick leave in postmenopausal women before diagnosis of hyperparathyroidism. In: BMJ (Clinical Research Ed.). Band 317, Nr. 7162, 26. September 1998, ISSN 0959-8138, S. 848–851, PMID 9748176, PMC 31094 (freier Volltext).
- P. Boudou, F. Ibrahim, C. Cormier, E. Sarfati, J. C. Souberbielle: A very high incidence of low 25 hydroxy-vitamin D serum concentration in a French population of patients with primary hyperparathyroidism. In: Journal of Endocrinological Investigation. Band 29, Nr. 6, Juni 2006, ISSN 0391-4097, S. 511–515, PMID 16840828.
- S. J. Silverberg, E. Shane, D. W. Dempster, J. P. Bilezikian: The effects of vitamin D insufficiency in patients with primary hyperparathyroidism. In: The American Journal of Medicine. Band 107, Nr. 6, Dezember 1999, ISSN 0002-9343, S. 561–567, PMID 10625024.
- Sundeep Khosla, Joseph Melton: Fracture risk in primary hyperparathyroidism. In: Journal of Bone and Mineral Research: The Official Journal of the American Society for Bone and Mineral Research. 17 Suppl 2, November 2002, ISSN 0884-0431, S. N103–N107, PMID 12412786.
- Wanjun Ren u. a.: Quiz page September 2008: progressive paraplegia in a long-term hemodialysis patient. Brown tumor compressing the thoracic spinal column. In: American Journal of Kidney Diseases. Band 52, Nr. 3, September 2008, ISSN 1523-6838, S. A37–A39, doi:10.1053/j.ajkd.2007.12.029 (ajkd.org [abgerufen am 7. November 2008]).
- Jane M Suh u. a.: Primary hyperparathyroidism: is there an increased prevalence of renal stone disease? In: AJR. American Journal of Roentgenology. Band 191, Nr. 3, September 2008, ISSN 1546-3141, S. 908–911, doi:10.2214/AJR.07.3160, PMID 18716127.
- E. Lundgren u. a.: Case-control study on symptoms and signs of "asymptomatic" primary hyperparathyroidism. In: Surgery. Band 124, Nr. 6, Dezember 1998, ISSN 0039-6060, S. 980–985; discussion 985–986, PMID 9854572.
- Theresia Weber u. a.: Effect of parathyroidectomy on quality of life and neuropsychological symptoms in primary hyperparathyroidism. In: World Journal of Surgery. Band 31, Nr. 6, Juni 2007, ISSN 0364-2313, S. 1202–1209, doi:10.1007/s00268-007-9006-6, PMID 17460812.
- L. Lind, S. Jacobsson, M. Palmér, H. Lithell, B. Wengle, S. Ljunghall: Cardiovascular risk factors in primary hyperparathyroidism: a 15-year follow-up of operated and unoperated cases. In: Journal of Internal Medicine. Band 230, Nr. 1, Juli 1991, ISSN 0954-6820, S. 29–35, PMID 2066709.
- Mark J Bolland, Andrew B Grey, Greg D Gamble, Ian R Reid: Association between primary hyperparathyroidism and increased body weight: a meta-analysis. In: The Journal of Clinical Endocrinology and Metabolism. Band 90, Nr. 3, März 2005, ISSN 0021-972X, S. 1525–1530, doi:10.1210/jc.2004-1891, PMID 15613408.
- M. Procopio, G. Magro, F. Cesario, A. Piovesan, A. Pia, N. Molineri, G. Borretta: The oral glucose tolerance test reveals a high frequency of both impaired glucose tolerance and undiagnosed Type 2 diabetes mellitus in primary hyperparathyroidism. In: Diabetic Medicine: A Journal of the British Diabetic Association. Band 19, Nr. 11, November 2002, ISSN 0742-3071, S. 958–961, PMID 12421435.
- Mishaela R Rubin, Shonni J Silverberg: Rheumatic manifestations of primary hyperparathyroidism and parathyroid hormone therapy. In: Current Rheumatology Reports. Band 4, Nr. 2, April 2002, ISSN 1523-3774, S. 179–185, PMID 11890884.
- M. Palmér H. O. Adami, U. B. Krusemo, S. Ljunghall: Increased risk of malignant diseases after surgery for primary hyperparathyroidism. A nationwide cohort study. In: American Journal of Epidemiology. Band 127, Nr. 5, Mai 1988, ISSN 0002-9262, S. 1031–1040, PMID 3358404.
- Inga-Lena Nilsson, Jan Zedenius, Li Yin, Anders Ekbom: The association between primary hyperparathyroidism and malignancy: nationwide cohort analysis on cancer incidence after parathyroidectomy. In: Endocrine-Related Cancer. Band 14, Nr. 1, März 2007, ISSN 1351-0088, S. 135–140, doi:10.1677/erc.1.01261, PMID 17395982.
- Karin B Michels, Fei Xue, Lena Brandt, Anders Ekbom: Hyperparathyroidism and subsequent incidence of breast cancer. In: International Journal of Cancer. Band 110, Nr. 3, 20. Juni 2004, ISSN 0020-7136, S. 449–451, PMID 15095313.
- T. Stefenelli u. a.: Cardiac abnormalities in patients with primary hyperparathyroidism: implications for follow-up. In: The Journal of Clinical Endocrinology and Metabolism. Band 82, Nr. 1, Januar 1997, ISSN 0021-972X, S. 106–112, PMID 8989242.
- Patrik Andersson, Erik Rydberg, Ronnie Willenheimer: Primary hyperparathyroidism and heart disease – a review. In: European Heart Journal. Band 25, Nr. 20, Oktober 2004, ISSN 0195-668X, S. 1776–1787, doi:10.1016/j.ehj.2004.07.010, PMID 15474692.
- J. C. Smith u. a.: Augmentation of central arterial pressure in mild primary hyperparathyroidism. In: The Journal of Clinical Endocrinology and Metabolism. Band 85, Nr. 10, Oktober 2000, ISSN 0021-972X, S. 3515–3519, PMID 11061493.
- Mishaela R Rubin u. a.: Arterial stiffness in mild primary hyperparathyroidism. In: The Journal of Clinical Endocrinology and Metabolism. Band 90, Nr. 6, Juni 2005, ISSN 0021-972X, S. 3326–3330, doi:10.1210/jc.2004-1400, PMID 15769995.
- Giorgio Coen: Calcimimetics, parathyroid hormone, and vascular calcification in chronic kidney disease. In: Kidney International. Band 74, Nr. 10, November 2008, ISSN 1523-1755, S. 1229–1231, doi:10.1038/ki.2008.417, PMID 18974757.
- Goce B. Spasovski: Bone health and vascular calcification relationships in chronic kidney disease. In: International Urology and Nephrology. Band 39, Nr. 4, 2007, ISSN 0301-1623, S. 1209–1216, doi:10.1007/s11255-007-9276-9, PMID 17899431.
- Hirotaka Komaba u. a.: Treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). In: Internal Medicine (Tokyo, Japan). Band 47, Nr. 11, 2008, ISSN 1349-7235, S. 989–994, doi:10.2169/internalmedicine.47.1051, PMID 18520108.
- M. D. Arenas u. a.: Management of calcific uremic arteriolopathy (calciphylaxis) with a combination of treatments, including hyperbaric oxygen therapy. In: Clinical Nephrology. Band 70, Nr. 3, September 2008, ISSN 0301-0430, S. 261–264, PMID 18793571.
- Hiroshi Sato u. a.: Primary hyperparathyroidism with duodenal ulcer and H. pylori infection. In: Internal Medicine (Tokyo, Japan). Band 41, Nr. 5, Mai 2002, ISSN 0918-2918, S. 377–380, PMID 12058887.
- R. A. Wermers u. a.: Incidence of primary hyperparathyroidism in Rochester, Minnesota, 1993–2001: an update on the changing epidemiology of the disease. In: J Bone Miner Res. Band 21, Nr. 1, 2006, S. 171–177, doi:10.1359/JBMR.050910, PMID 16355286.
- G. I. Salti, I. Fedorak, T. Yashiro, N. Fulton, H. Hara, D. Yousefzadeh, E. L. Kaplan: Continuing evolution in the operative management of primary hyperparathyroidism. In: Archives of Surgery. Band 127, Nr. 7, Juli 1992, ISSN 0004-0010, S. 831–836; discussion 836–837, PMID 1524484.
- D. Bartsch, C. Nies, C. Hasse, J. Willuhn, M. Rothmund: Clinical and surgical aspects of double adenoma in patients with primary hyperparathyroidism. In: British Journal of Surgery. Band 82, Nr. 7, Juli 1995, ISSN 0007-1323, S. 926–929, PMID 7648110.
- Alexander Maret, Isabelle Bourdeau, Changlin Ding, Shrihari S Kadkol, William H Westra, Michael A Levine: Expression of GCMB by intrathymic parathyroid hormone-secreting adenomas indicates their parathyroid cell origin. In: The Journal of Clinical Endocrinology and Metabolism. Band 89, Nr. 1, Januar 2004, ISSN 0021-972X, S. 8–12, PMID 14715818.
- James M Ruda, Christopher S Hollenbeak, Brendan C Stack: A systematic review of the diagnosis and treatment of primary hyperparathyroidism from 1995 to 2003. In: Otolaryngology – Head and Neck Surgery. Band 132, Nr. 3, März 2005, ISSN 0194-5998, S. 359–372, doi:10.1016/j.otohns.2004.10.005, PMID 15746845.
- A. G. Wynne, J. van Heerden, J. A. Carney, L. A. Fitzpatrick: Parathyroid carcinoma: clinical and pathologic features in 43 patients. In: Medicine. Band 71, Nr. 4, Juli 1992, ISSN 0025-7974, S. 197–205, PMID 1518393.
- Wanjun Ren, Xiaoping Wang, Bin Zhu, Zidong Liu: Quiz page September 2008: progressive paraplegia in a long-term hemodialysis patient. Brown tumor compressing the thoracic spinal column. In: American Journal of Kidney Diseases: The Official Journal of the National Kidney Foundation. Band 52, Nr. 3, September 2008, ISSN 1523-6838, S. A37–A39, doi:10.1053/j.ajkd.2007.12.029 (ajkd.org [abgerufen am 7. November 2008]).
- George M. Nassar, Juan Carlos Ayus: Brown Tumor in End-Stage Renal Disease. In: N Engl J Med. Band 341, Nr. 22, 25. November 1999, S. 1652, doi:10.1056/NEJM199911253412204.
- A. N. Hollenberg, A. Arnold: Hypercalcemia with low-normal serum intact PTH: a novel presentation of primary hyperparathyroidism. In: Am J Med. Band 91, Nr. 5, November 1991, S. 547–548, PMID 1951417.
- S. R. Nussbaum u. a.: Highly sensitive two-site immunoradiometric assay of parathyrin, and its clinical utility in evaluating patients with hypercalcemia. In: Clin Chem. Band 33, Nr. 8, August 1987, S. 1364–1367, PMID 3608153 (clinchem.org).
- W. Millesi, B. Niederle u. a.: Osteolysen im Kieferbereich als erster Hinweis auf einen primären Hyperparathyreoidismus. In: Acta Chirurgica Austriaca. Volume 26, Issue 6, 1994, S. 410–414. doi:10.1007/BF02620046
- Gel-H Fuleihan: Familial benign hypocalciuric hypercalcemia. In: J Bone Miner Res. 17 Suppl 2, 2002, S. 51–56, PMID 12412778.
- H. Lowe: Normocalcemic primary hyperparathyroidism: further characterization of a new clinical phenotype. In: J Clin Endocrinol Metab. Nr. 92, 2007, S. 3001, PMID 17536001 (endojournals.org).
- P. Ureña u. a.: Bone mineral density, biochemical markers and skeletal fractures in haemodialysis patients. In: Nephrol Dial Transplant. Nr. 18, 2003, S. 2325–2331, PMID 14551361 (oxfordjournals.org).
- S. A. Jamal u. a.: Low bone mineral density and fractures in long-term hemodialysis patients: a meta-analysis. In: Am J Kidney Dis. Band 49, Nr. 5, Mai 2007, S. 674–681, PMID 17472850.
- Elena Ambrogini u. a.: Surgery or Surveillance for Mild Asymptomatic Primary Hyperparathyroidism: A Prospective, Randomized Clinical Trial. In: J Clin Endocrinol Metab. Nr. 92, 2007, S. 3114–3121 (Abstract).
- Lindi H. VanderWalde u. a.: The Effect of Parathyroidectomy on Bone Fracture Risk in Patients With Primary Hyperparathyroidism. In: Arch Surg. Nr. 141, 2006, S. 885–891 (Abstract).
- John P. Bilezikian u. a.: Summary Statement from a Workshop on Asymptomatic Primary Hyperparathyroidism: A Perspective for the 21st Century. In: J. Clin. Endocrinol. Metab. Nr. 12, 2002, S. 5353–5361 (Artikel).
- B. Farford, R. J. Presutti, T. J. Moraghan: Nonsurgical Management of Primary Hyperparathyroidism. In: Mayo Clin Proc. Band 82, Nr. 3, 2007, S. 351–355.
- Eleanor Lederer, Rosemary Ouseph: Chronic Kidney Disease. In: American Journal of Kidney Diseases. Band 49, Nr. 1, 2007, S. 162–171 (Artikel).
- K/DOQI: Clinical Practice Guidelines for Bone Metabolism and Disease in Chronic Kidney Disease – Guideline 14. In: American Journal of Kidney Diseases. 42, Supplement 3, 2003, S. 1–202 (Artikel (Memento vom 21. Februar 2013 im Internet Archive)). Clinical Practice Guidelines for Bone Metabolism and Disease in Chronic Kidney Disease – Guideline 14 (Memento des Originals vom 21. Februar 2013 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis.
- K/DOQI: Clinical Practice Guidelines for Bone Metabolism and Disease in Chronic Kidney Disease Guideline 15. In: American Journal of Kidney Diseases. Band 42, Supplement 3, Oktober 2003, S. 1–202 (Artikel (Memento vom 20. Juni 2012 im Internet Archive)). Clinical Practice Guidelines for Bone Metabolism and Disease in Chronic Kidney Disease Guideline 15 (Memento des Originals vom 20. Juni 2012 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis.