Multiple endokrine Neoplasie
Multiple endokrine Neoplasie (Abkürzung: MEN) ist der Überbegriff für verschiedene spezifische erbliche Tumorerkrankungen, die eine krebsartige Wucherung endokriner Drüsen begünstigen und mit Überfunktionssyndromen einhergehen. Die Multiple endokrine Neoplasie hat in der Bevölkerung eine Häufigkeit von 1:50.000.
| Klassifikation nach ICD-10 | |
|---|---|
| D44 | Neubildung unsicheren oder unbekannten Verhaltens der endokrinen Drüsen |
| D44.8 | Beteiligung mehrerer endokriner Drüsen Multiple endokrine Adenomatose |
| ICD-10 online (WHO-Version 2019) | |
Sie wird in drei verschiedene Typen eingeteilt: MEN 1, MEN 2A und MEN 2B.
MEN 1
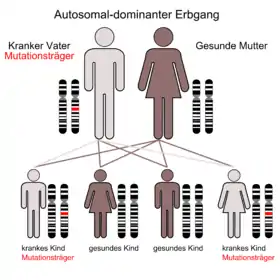
Die MEN 1 (Wermer-Syndrom, nach Paul Wermer, amerikanischer Internist (1898–1975)[1]) ist durch eine Neoplasie der Nebenschilddrüsen, der Hypophyse und der Inselzellen der Bauchspeicheldrüse gekennzeichnet. Das Krankheitsbild wird autosomal-dominant vererbt, das heißt Kinder eines Betroffenen haben eine 50-prozentige Wahrscheinlichkeit, das betreffende Gen zu erwerben und letztlich an einem oder mehreren der verbundenen Tumoren zu erkranken.
Obwohl jeder einzelne der entstehenden Tumoren klonalen Ursprungs ist, das heißt aus jeweils einer einzelnen entarteten Zelle hervorgegangen ist, tragen doch alle Zellen des betroffenen Patienten den genetischen Fehler. Daher ist immer mit einer Tumormanifestation an anderen Lokalisationen zu rechnen. Die Manifestationen der MEN 1 entwickeln sich über drei bis vier Jahrzehnte, die festgestellten tatsächlichen Tumoren hängen vom Zeitpunkt der Diagnosestellung ab.
Die häufigste Manifestation des MEN-1-Syndroms ist der Hyperparathyreoidismus; bis zum 40. Lebensjahr ist bei der Mehrzahl der Genträger die Krankheit zum Ausbruch gekommen. Typische Symptome sind kalziumhaltige Nierensteine, Knochenveränderungen sowie gastrointestinale und muskuläre Beschwerden. Die zugrundeliegende Hyperkalzämie tritt dagegen meist schon im Jugendalter auf.
Die zweithäufigste Manifestation sind Neoplasien der Inselzellen im Pankreas. Sie treten meistens gemeinsam mit dem Hyperparathyreoidismus auf. Die häufigsten Hormone, die aus den Inselzellen verstärkt freigesetzt werden, sind:
| Häufigkeit | vermehrt produziertes Hormon | klinisches Syndrom |
|---|---|---|
| 75–85 % | pankreatisches Polypeptid | |
| 60 % | Gastrin | Zollinger-Ellison-Syndrom |
| 30–35 % | Insulin | Insulinom |
| 3–5 % | vasoaktives intestinales Peptid (VIP) | Verner-Morrison-Syndrom |
| 5–10 % | Glucagon | Glucagonom |
| 1–5 % | Somatostatin |
Seltener gebildete Hormone sind ACTH, CRH, GHRH, Calcitoninprodukte, Neurotensin, Gastroinhibitorisches Peptid und weitere.
Menin, das Protein, das beim MEN1-Syndrom krankhaft verändert ist, nimmt im normalen Stoffwechsel an Entwicklungsprozessen und der DNA-Reparatur teil. Aktuell sind etwa 400 unterschiedliche Mutationen auf dem MEN1-Gen bekannt.
MEN 2
Die MEN-2-Syndrome sind durch medulläre Schilddrüsenkarzinome und Phäochromozytome gekennzeichnet.
Ursächlich sind aktivierende Mutationen des RET-Protoonkogens. Das Gen befindet sich auf dem langen Arm von Chromosom 10 (10q11.2) und codiert für einen Tyrosinkinaserezeptor. Die Erkrankung wird autosomal-dominant vererbt. Es sind mehrere verschiedene Mutationsarten bekannt, die zur Ausbildung des Syndroms führen können. Gerade beim familiär gehäuften Auftreten von C-Zellkarzinomen liegt der Verdacht einer MEN 2 nahe.
MEN 2
Das MEN-2-Syndrom wurde 1961 von J. H. Sipple beschrieben und wird daher auch als Sipple-Syndrom bezeichnet.[2]
Die häufigste Manifestation ist das medulläre Schilddrüsenkarzinom, das meist bereits im Kindesalter auftritt und mit einer Hyperplasie der C-Zellen beginnt, die Calcitonin produzieren.
Ein Phäochromozytom kommt bei etwa 50 % der Patienten vor. In der Hälfte der Fälle tritt der Tumor beidseitig auf, mehr als die Hälfte der Patienten entwickelt nach einseitiger Entfernung der Nebenniere innerhalb von 10 Jahren ein Phäochromozytom der anderen Seite.
Bei 15 bis 20 % der Patienten tritt ein primärer Hyperparathyreoidismus auf, meist zwischen dem 20. und 40. Lebensjahr.
Das MEN-2A-Syndrom hat noch folgende Sonderformen:
- Familiäres Medulläres Schilddrüsenkarzinom
Auch FMTC (engl. familial medullary thyroid carcinoma)
- MEN 2A mit kutaner Lichenamyloidose
- MEN 2A mit Morbus Hirschsprung
MEN 3
Das MEN-3-Syndrom (ehemals MEN-2B-Syndrom) wird auch als Williams-Pollok-Syndrom oder Gorlin-Vickers-Syndrom bezeichnet.[3] Es umfasst neben dem Medullären Schilddrüsenkarzinom und Phäochromozytom Neurinome der Schleimhäute, Ganglioneuromatose, und einen marfanoiden Habitus (Körperbau ähnlich wie beim Marfan-Syndrom: schlanker Körperbau, lange Extremitäten, Arachnodaktylie, Überstreckbarkeit der Gelenke).
Das Medulläre Schilddrüsenkarzinom tritt bei dieser Form früher auf und wächst aggressiver als beim MEN 2A. Die Erkrankung tritt gelegentlich schon im 1. Lebensjahr mit Metastasen auf und endet meist im 2. oder 3. Lebensjahrzehnt tödlich.
Charakteristisch sind die Schleimhautneurinome, die an der Zungenspitze, den Augenlidern und im gesamten Magen-Darm-Trakt vorkommen.
Früherkennung und Therapie
Für die Prognose der Patienten ist der Verlauf des Medullären Schilddrüsenkarzinoms entscheidend. Der letztlich tödliche Verlauf lässt sich nur durch eine Thyreoidektomie (vollständige Entfernung der Schilddrüse) vor dem Auftreten von Metastasen verhindern. Ein Screening der Familienangehörigen eines Patienten mit MEN 2A ist mittels entsprechender DNA-Analyse leicht möglich, wenn die entsprechende Mutation des RET-Protoonkogens des Indexpatienten bekannt ist.
Bei Kindern, bei denen eine Mutation mit hohem Entartungsrisiko gefunden wurde, oder eine Mutation, die mit einem MEN 2B assoziiert ist, wird unmittelbar nach Diagnose-Stellung die Thyreoidektomie und zentrale Halslymphknoten-Dissektion durchgeführt. Bei bestimmten anderen Mutationen mit mittlerem Entartungsrisiko wird die Entfernung der Schilddrüse zumindest vor dem 6. Lebensjahr empfohlen. Bei Mutationen mit geringem Risiko wird die Operation zwischen 6. und 12. Lebensjahr empfohlen. Der früher auch empfohlene ein- bis zweijährliche Pentagastrin-Test (Bestimmung des Serum-Calcitonins nach Stimulation mit Pentagastrin) wird aktuell (2015) nicht mehr durchgeführt, weil Pentagastrin nicht mehr verfügbar ist.
Bei allen nachgewiesenen Mutationen im Bereich des RET-Protoonkogens sollten jährliche Screening-Untersuchungen auf das Vorliegen eines Phäochromozytoms durchgeführt werden, hierfür werden die Katecholamine und Metanephrine im Blutplasma oder Sammelurin bestimmt.
Ein Hyperparathyreoidismus lässt sich durch Bestimmung des Serum-Kalziums und des Parathormons in mehrjährigen Abständen erkennen, bei familiärer Häufung in kürzeren Abständen.
Literatur
- Dietel, Dudenhausen, Suttorp (Hrsg.): Harrisons Innere Medizin. Berlin 2003, ISBN 3-936072-10-8, S. 2387–2391.
- F. Marini, A. Falchetti, F. Del Monte, S. Carbonell Sala, I. Tognarini, E. Luzi, M. L. Brandi: Multiple endocrine neoplasia type 2. In: Orphanet J Rare Dis. 2006 Nov 14;1, S. 45. PMID 17105651, PMC 1654141 (freier Volltext)