Phäochromozytom
Das Phäochromozytom (von griechisch phaios „dunkel“, chroma „Farbe“, neulateinisch zytus „Zelle“, -om „Tumor“) ist eine Erkrankung der chromaffinen Zellen des Nebennierenmarks mit einer Inzidenz von 1/100.000 Personen/Jahr. Es handelt sich um einen Tumor, der Katecholamine produziert (Noradrenalin, Adrenalin und Metanephrine), die einen Bluthochdruck verursachen. Maligne Phäochromozytome bilden auch Dopamin. Er ist zu 85 % im Nebennierenmark lokalisiert, kommt jedoch auch in den Nervengeweben des thorakalen und abdominalen Grenzstrangs vor (Paragangliom), produziert hier aber fast ausschließlich Noradrenalin. Sehr selten tritt das Phäochromozytom zum Beispiel in der Harnblase[1][2] und anderen aus Neuralleistenzellen abgeleiteten Geweben auf. Etwa 10 % der Phäochromozytome sind maligne, hierzu finden sich in der Literatur unterschiedliche Prozentangaben.
| Klassifikation nach ICD-10 | |
|---|---|
| C74.1 | Bösartige Neubildung des Nebennierenmarks |
| D35.0 | Gutartige Neubildung der Nebenniere |
| D44.1 | Neubildung der Nebenniere unsicheren oder unbekannten Verhaltens |
| E27.5 | Nebennierenmarküberfunktion |
| ICD-10 online (WHO-Version 2019) | |
_histopathology.jpg.webp)
Das Phäochromozytom tritt isoliert oder im Rahmen eines MEN-2-Syndroms (multiple endokrine Neoplasien), beim von-Hippel-Lindau-Syndrom Typ 2 und bei der Neurofibromatose Typ 1 (Morbus Recklinghausen) auf.
Erstbeschreibung
Die erste Beschreibung eines Phäochromozytoms geht auf Felix Fränkel 1886 zurück, der den Fall der 18-jährigen Minna Roll aus Wittenweier wiedergab, die innerhalb von zehn Tagen in der Universitätsklinik Freiburg starb. Pathologisch wurde an beiden Nieren ein Nebennieren- und Angiosarkom beschrieben. Bei der Präparate-Herstellung wurde die Chromat-haltige Fixationslösung nach Müller verwendet, die ausschließlich die Tumorzellen bräunlich anfärbte, was 1912 vom Berliner Pathologen Ludwig Pick als Chromatbraun, oder phäo chrom (griech.) bezeichnet wurde. Er verwendete als erster den Begriff Phäochromozytom. In einer neueren Studie konnte anhand überlebender Verwandter der ersten Patientin nachgewiesen werden, dass sie an einer Multiplen endokrinen Neoplasie MEN-2 litt.[3]
Symptome
Der Patient klagt vor allem über einen (durch Ausschüttung von Stresshormonen bedingten) anfallsartigen Bluthochdruck (paroxysmale Hypertonie) oder dauerhafte Blutdruckerhöhung (persistierende Hypertonie, oft bei Kindern). Während der Phasen des erhöhten Blutdruckes treten Kopfschmerzen, Schwindel, Herzrasen und Schwitzen auf. Weitere Zeichen sind blasse Haut, Blutzuckererhöhung (Hyperglykämie), eine Leukozytose und Gewichtsverlust. Anfallsweise können Ohnmacht, Übelkeit, Erbrechen, vermehrte Tränen- und Speichelbildung auftreten.[4]
Diagnose
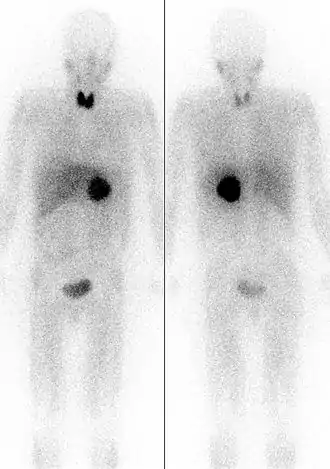
Links: von vorne. Rechts: von hinten.
Die Diagnose basiert auf den verdächtigen klinischen Symptomen. Besonders verdächtig sind sporadisch auftretende Bluthochdruckattacken, bei der die gängigen medikamentösen Therapien nicht ansprechen. Da ein Phäochromozytom für den betroffenen Patienten eine sehr ernsthafte Gefahr darstellt, muss eine zuverlässige Labordiagnostik erfolgen.
Für die laborchemische Untersuchung stehen eine Reihe von Verfahren zur Verfügung. Dies sind in erster Linie die Bestimmung der Katecholamine beziehungsweise deren Abbauprodukte aus Urin oder Plasma. Die Bestimmung der Vanillinmandelsäure ist wegen der geringen Sensitivität heute nicht mehr angezeigt. Besser ist die Bestimmung der Katecholamine (Adrenalin, Noradrenalin) aus dem 24-Stunden-Sammelurin oder aus Plasmaproben oder die Bestimmung der Metanephrine (Metanephrin und Normetanephrin) aus dem 24-Stunden-Urin. Der Nachteil der vorgenannten Labormethoden ist jedoch, dass weder ein positives noch ein negatives Ergebnis eine sichere Aussage ermöglicht.
Dies ist lediglich durch die quantitative Bestimmung der freien Metanephrine im Plasma möglich. Wenn diese nicht erhöht sind, ist ein Phäochromozytom mit Sicherheit auszuschließen. Die diagnostische Sensitivität dieser Methode ist nahezu 100 %, was in mehreren internationalen Studien gezeigt werden konnte.[5]
Wenn die Metanephrine im Plasma dagegen auch im wiederholten Falle erhöht sind, besteht ein starker Verdacht auf ein Phäochromozytom. Seit jüngster Zeit ist die quantitative Bestimmung der freien Plasma-Metanephrine mit einer Routinebestimmung (RIA- bzw. ELISA Methode) in jedem gut ausgerüsteten Labor durchführbar. Die Plasmaentnahme darf dabei erst nach einer 20-minütigen Ruhephase im Liegen durchgeführt werden, da es sonst zu viele falsch-positive Ergebnisse gibt.
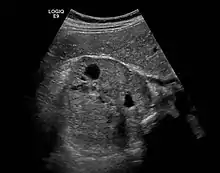
Wenn die Bestimmung der Plasma-Metanephrine wiederholt ein positives Ergebnis bringt, muss eine weiterführende Lokalisationsdiagnostik durchgeführt werden. Dies geschieht mittels bildgebender Verfahren wie Computertomographie und Sonographie oder MRT-Bildgebung. Bei der CT ist zu berücksichtigen, dass beim Einlaufen von Jod-Kontrastmittel Katecholamine ausgeschüttet werden können, was bei der MRT nicht der Fall ist. Die nuklearmedizinische Methode der MIBG-Szintigrafie (Metaiodobenzylguanidin) dient vor allem dem Ausschluss von Phäochromozytomen außerhalb der Nebenniere. Diese Substanz lagert sich vornehmlich in den betroffenen chromaffinen Zellen des Phäochromozytoms ab. Die neueste und zuverlässigste Form der nuklearmedizinischen Methode ist bei Phäochromzytomen das so genannte DOPA-PET. Es wird zurzeit (Stand 2012) in Deutschland allerdings lediglich in wenigen Zentren angeboten. Zusätzlich muss auf alle Fälle nach Tumoren eines eventuellen MEN-Syndroms gesucht werden.
Differenzialdiagnose
Differentialdiagnostisch müssen andere Tumoren der Nebenniere (z. B. Ganglioneurom) ausgeschlossen werden. Bei 98 % der Patienten mit anfallsweisem Bluthochdruck kann ein Phäochromozytom ausgeschlossen werden. Häufig liegt dann eine schwere paroxysmale arterielle Hypertonie vor, die auch als Pseudophäochromozytom bezeichnet wird.
Eine Vielzahl weiterer Krankheitsbilder kann zu anfallsartigem Bluthochdruck führen und muss in die Differenzialdiagnose mit einbezogen werden: Angststörung, Panikstörung, Hyperthyreose, beta-adrenerge hyperdyname Zirkulationsstörung, Cluster-Kopfschmerz, Migräne, hypertensive Enzephalopathie, koronare Herzkrankheit, Nierenarterienstenose, Schlaganfall, Gehirntumor, Gehirnverletzung, Gehirnblutung, Kompression der Medulla oblongata, Anfallsleiden, Karzinoid, Drogen wie Kokain, Mutterkornalkaloide, Diethylamin, Amphetamine, Tyrosin in Kombination mit Monoaminooxidase-Hemmern, gestörter Barorezeptor-Reflex und Münchhausen-Syndrom.[6]
Therapie
Die chirurgische Therapie erfolgt durch die operative Resektion des Tumors. Mindestens eine Woche vor dem Eingriff wird eine Senkung des Blutdruckes mit einem Alpha-Blocker (z. B. Phenoxybenzamin) begonnen. Danach gibt man einen unselektiven Beta-Blocker dazu. Diese Gabe findet statt, da bei der operativen Entfernung des Tumors große Mengen Katecholamine freigesetzt werden, die ohne entsprechende Blocker lebensgefährlich sein könnten. Die Alpha-Blocker müssen in jedem Fall vor den Betablockern gegeben werden, da sonst eine hypertensive Krise droht. Falls bei einem Phäochromozytom zuerst Beta-Blocker gegeben werden, fällt die über den β2-Rezeptor vermittelte vasodilatatorische Wirkung weg und es kommt zum Blutdruckanstieg (Stimulation der α1-Rezeptoren bewirkt eine Vasokonstriktion).
Bei einem einseitigen Phäochromozytom führt man eine totale Entnahme der Nebenniere dieser Seite durch, im Rahmen eines MEN-Syndroms entfernt man beidseitig lediglich das Nebennierenmark. Bei 80 % der Patienten normalisieren sich postoperativ Katecholaminspiegel und Blutdruck.
Bei Metastasenbildung führt man eine systemische Radionuklidtherapie (MIBG-Therapie) oder eine Polychemotherapie[7] durch. Weitere Optionen sind die Gabe von Interferonen und Octreotid.
Literatur
- Graeme Eisenhofer: Biochemical Diagnosis of Pheochromocytoma – Is it Time to Switch to Plasma-Free Metanephrines? In: J Clin Endocrinol Metab, 88 (2), S. 550–552, PMID 12574178.
- Heinz Lüllmann et al.: Pharmakologie und Toxikologie. 16. Auflage, Thieme Verlag, Stuttgart 2006, ISBN 3-13-368516-3.
- Lois Jovanovic, Genell J. Subak-Sharpe: Hormone. Das medizinische Handbuch für Frauen. (Originalausgabe: Hormones. The Woman’s Answerbook. Atheneum, New York 1987) Aus dem Amerikanischen von Margaret Auer, Kabel, Hamburg 1989, ISBN 3-8225-0100-X, S. 319 f. und 383.
Weblinks
- MSD-Manual. Ausgabe für medizinische Fachkreise: Phäochromozytom.
- Weitergehende Literatur: www.pheochromocytoma.org
Einzelnachweise
- H. Lipsky, G. Leitner: Das Phäochromozytom der Harnblase. In: Aktuelle Urologie. 20, 1989, S. 342, doi:10.1055/s-2008-1061238.
- S. Vyas, N. Kalra, S. K. Singh, M. M. Agarwal, A. K. Mandal, N. Khandelwal: Pheochromocytoma of urinary bladder. In: Indian journal of nephrology. Band 21, Nummer 3, Juli 2011, S. 198–200, doi:10.4103/0971-4065.78072, PMID 21886982, PMC 3161440 (freier Volltext).
- H. P. H. Neumann et al.: Evidence of MEN-2 in the original description of classic pheochromocytoma. In: New England Journal of Medicine, 2007, 57 (13), S. 1311
- Carsten Kunz, Winfried Kunz, Mechthild Seel: Kompaktwissen Krankenpflege. Schlütersche, 2004, ISBN 978-3-87706-759-8, S. 345.
- J. W. Lenders, K. Pacak, M. M. Walther, W. M. Linehan, M. Mannelli, P. Friberg, H. R. Keiser, D. S. Goldstein, G. Eisenhofer: Biochemical diagnosis of pheochromocytoma: which test is best? In: Journal of the American Medical Association. Band 287, Nummer 11, März 2002, S. 1427–1434, PMID 11903030. Volltext (PDF).
- S. J. Mann: Severe paroxysmal hypertension (pseudopheochromocytoma): understanding the cause and treatment. In: Archives of internal medicine. Band 159, Nummer 7, April 1999, S. 670–674, PMID 10218745 (Review). PDF.
- Tim Scholz et al.: Current Treatment of Malignant Pheochromocytoma. In: The Journal of Clinical Endocrinology & Metabolism, 2007, Vol. 92, No. 4, S. 1217–1225.