Neurofibromatose Typ 1
Die Neurofibromatose Typ 1 (kurz: NF1), auch Von-Recklinghausen-Krankheit, Morbus Recklinghausen, kurz auch Neurofibromatose Recklinghausen (benannt nach ihrem Entdecker Friedrich Daniel von Recklinghausen) oder periphere Neurofibromatose, ist eine autosomal-dominant und monogen vererbte Multiorganerkrankung, bei der es zu multiplen Neurofibromen peripherer Nerven und zu Hautmissbildungen kommt.
| Klassifikation nach ICD-10 | |
|---|---|
| Q85.0 | Neurofibromatose (nicht bösartig) - von-Recklinghausen-Krankheit |
| ICD-10 online (WHO-Version 2019) | |
Etwa 50 Prozent der Betroffenen haben eine Neumutation. NF1 betrifft vor allem Haut und Nervensystem. Sie wird daher den neurokutanen Erkrankungen (Phakomatosen) zugeordnet. Typische Veränderungen an der Haut sind Café-au-lait-Flecken sowie Neurofibrome. Als Café-au-lait-Flecken bezeichnet man milchkaffeefarbene Hyperpigmentierungen der Haut. Sie liegen im Niveau der Haut, können bei allen Menschen auftreten und sind harmlos. Bei Menschen mit einer NF1 treten sie gehäuft auf. Als Neurofibrome bezeichnet man gutartige Tumoren, die von den Zellen der Schwann’schen Scheiden kleiner in der Haut verlaufender Nervenfasern ausgehen. Im zentralen Nervensystem (ZNS) treten gehäuft Tumoren verschiedener Lokalisation auf. Patienten können aufgrund der Erkrankung geistig behindert (minderbegabt) sein und an epileptischen Anfällen leiden. Des Weiteren sind häufig Augen (mit resultierender Sehschwäche) und Knochen mitbetroffen. Die Pubertät kann verfrüht oder verspätet eintreten. Eine Neurofibromatose wird durch eine Veränderung in einem Gen hervorgerufen, welches normalerweise hemmend auf die Zellteilung Einfluss nimmt. Es kommt daher zu überschießender Gewebsvermehrung und damit zu den typischen Veränderungen. Die Diagnose wird meist anhand des klinischen Bildes bereits in der Kindheit gestellt. Da es sich bei Morbus Recklinghausen um eine genetische Erkrankung handelt, ist eine Therapie, welche ihre Ursache beseitigt, derzeit nicht möglich. Es werden daher nur Veränderungen behandelt, die für den Patienten störend oder gefährlich sind. Eine weitere bekannte Form der Neurofibromatose ist die Neurofibromatose Typ 2 (NF2), welche wesentlich seltener auftritt und von einer Mutation auf einem anderen Gen verursacht wird.
Geschichte
Eine eher anekdotische Erstbeschreibung findet sich bei Robert William Smith 1849. Friedrich Daniel von Recklinghausen legte 1882 die erste präzise klinische und pathologische Charakterisierung vor. Alex Thomsen gab um 1900 die ersten statistischen Daten und eine ausführliche Bibliographie heraus. Joseph Merrick, der sogenannte „Elefantenmensch“, galt lange Zeit als ein Beispiel für die entstellenden Auswirkungen der Recklinghausenschen Krankheit. Sein Leben im viktorianischen England war Grundlage für Bücher und Filme, insbesondere David Lynchs Der Elefantenmensch. Merricks schwere Entstellungen prägten die weitverbreiteten falschen Vorstellungen von der Monstrosität der Patienten mit einer Neurofibromatose Typ 1. Nach einer DNA-Analyse im Jahre 2003 litt Merrick aber am Proteus-Syndrom, wobei eine zusätzliche Erkrankung an Neurofibromatose Typ 1 wahrscheinlich ist. Die ältesten noch erhaltenen schriftlichen Schilderungen über die Erkrankung stammen aus dem 13. Jahrhundert.
Inzidenz, Erbgang und Epidemiologie
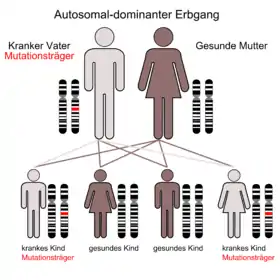
Man schätzt etwa 30 bis 40 Erkrankte auf 100.000 Einwohner, was einer Erwartung von einem betroffenen Kind pro 2500 bis 3300 Geburten entspricht. In der Hälfte der Fälle geht man davon aus, dass eine Neumutation zu den Veränderungen im Erbgut führt. Alle bisherigen Beobachtungen bestätigen den autosomal dominanten Erbgang, was bedeutet, dass ein betroffener Elternteil mit einer Wahrscheinlichkeit von 50 Prozent die Erkrankung an seine Kinder weitergibt. Man findet keine unterschiedlichen Häufigkeiten in verschiedenen Regionen der Erde oder unter Angehörigen anderer ethnischer Gruppen. Allerdings erkranken Männer etwas häufiger als Frauen. Die hohe Rate der Neumutation wird mit dem Umstand erklärt, dass das NF-1-Gen sehr groß ist und somit viel Angriffsfläche für genetische Veränderungen bietet.[1]
Pathogenese und Molekularbiologie
Die Neurofibromatose Typ 1 war eine der ersten erblichen Tumorerkrankungen, deren Genetik aufgeklärt werden konnte.[2] Der Neurofibromatose-Typ-1-Lokus liegt auf dem Chromosom 17 Genlocus q11.2.[3] Er ist komplex und kodiert möglicherweise für ein den intrazellulären Signalpfad modulierendes Protein. Der Neurofibromatose-Typ-1-Genlocus umspannt ca. 400.000 Basenpaare. In einem mehr als 40.000 Basenpaare großen Intron dieses Locus finden sich drei Gene in entgegengesetzter Leserichtung: OMPG codiert für ein membrangebundenes Glykoprotein des Oligodendrozyten-Myelins, EVI2A und EVI2B codieren für virale Insertionssequenzen. Am Neurofibromatose-Typ-1-Lokus sind Translokationen (1,17) und (17,22), Deletionen, Insertionen und Punktmutationen beschrieben.[4] Die über 50 Exons des Gens kodieren für verschiedene ca. 9 bis 11 kB große Transkripte.[5] Ein ca. 7800 Basenpaare umfassender open reading frame des genomischen Lokus erlaubt die Ableitung eines Proteins mit ca. 2500 Aminosäuren. Das Neurofibromatose-Typ-1-Peptid (Neurofibromin) zeigt Sequenz-Homologien mit dem von Säugetieren bekannten GAP (GTPase aktivierendes Protein)[6] und den IRA1- und IRA2-Genen der Hefe.[7] Die GAP-verwandte Domäne des NF-1-Peptids bindet in vitro an das „ras p21“-Protein.[8] Die katalytische Domäne von NF-1 stimuliert die GTPase-Aktivität von ras p21.[9] Wenn GTPasen durch ihr (individuelles) GAP aktiviert werden, dann hydrolysieren sie das gebundene GTP zu GDP und sind als solche nicht mehr in der Lage, ihren Effektor zu stimulieren. Dieser Effektor ist im Falle von ras p21 ein über den Phosphatidylinositol-Pfad vermitteltes mitogenes (die Zellteilung stimulierendes) Signal. Defekte GAPs können somit ein mitogenes Signal nicht mehr abschalten, die Zellen proliferieren unkontrolliert.
Das Mikrodeletionssyndrom 17q11.2 kann als Sonderform dieser Neurofibromatose Typ 1 angesehen werden.
Pathologie
Tumoren
Bei der Neurofibromatose kommen eine Reihe von Tumoren, die sowohl das zentrale Nervensystem betreffen als auch außerhalb davon auftreten können, gehäuft vor.[10]
Tumoren des Nervensystems
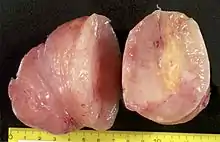
Neurofibrome
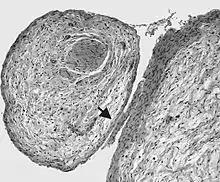
Für die Erkrankung besonders charakteristisch ist das Auftreten von Neurofibromen, bei denen es sich im Gegensatz zu sporadisch auftretenden Neurofibromen häufig um Neurofibrome der Haut (dermale Neurofibrome) oder sogenannte plexiforme Neurofibrome handelt. Dermale Neurofibrome sind gutartige, gut abgrenzbare, unter der Haut gelegene, von kleinen Hautnervenästen ausgehende Tumoren, die aus Schwann-Zellen und Fibroblasten-ähnlichen Zellen bestehen. Plexiforme Neurofibrome infiltrieren diffus größere Nervenäste und führen so zu einer kolbenförmigen Auftreibung. Im Gegensatz zu gewöhnlichen Neurofibromen ist das Risiko einer bösartigen Entartung mit etwa zehn Prozent deutlich erhöht.
Bösartige periphere Nervenscheidentumoren
Bösartige periphere Nervenscheidentumoren treten bei der Neurofibromatose bereits im jüngeren Lebensalter auf und können histologisch an Skelettmuskulatur erinnernde rhabdomyoblastische oder drüsenähnliche glanduläre Elemente enthalten. Solche Tumoren, die auch als Triton-Tumor bezeichnet werden, sind hochcharakteristisch für die Neurofibromatose Typ 1.
Gliome
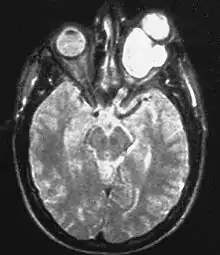
Den Großteil der bei Neurofibromatose Typ 1 auftretenden Gliome machen die im Bereich des Sehnervs (Nervus opticus) lokalisierten gutartigen pilozytischen Astrozytome aus, die bei dieser Lokalisation auch als Optikusgliome bezeichnet werden. Bei der Neurofibromatose Typ 1 treten Optikusgliome charakteristischerweise bilateral auf und betreffen so beide Sehnerven. Optikusgliome können bei Patienten mit Neurofibromatose Typ 1 einen über viele Jahre statischen Verlauf haben. Andere Gliome, die bei Neurofibromatose Typ 1 vermehrt auftreten, sind diffuse Astrozytome und das bösartige Glioblastom.
Tumoren außerhalb des Nervensystems
Das Auftreten von Phäochromozytomen, Tumoren des Nebennierenmarks, ist erhöht.[11] Dasselbe gilt für andere seltene Tumoren wie Rhabdomyosarkome, das juvenile Xanthogranulom, gastrointestinale Stromatumoren (GIST), medulläre Schilddrüsenkarzinome sowie die Chronische myelomonozytäre Leukämie. Im Bereich des Halses und des Brustkorbs auftretende mediastinale, atemwegsnahe Tumoren können zu schweren Störungen der Atmung[12] führen.
Pigmentstörungen
Café-au-lait-Flecken, Freckling im Bereich beider Achselhöhlen (sogenanntes Axillary Freckling) und Lisch-Knötchen gehen auf Veränderungen der Melanozyten der Haut zurück. Dabei sind Café-au-lait-Flecken oft schon bei der Geburt vorhanden und werden im Lauf der Kindheit häufiger und größer. Freckling, eine Art Sommersprossenbildung im Bereich der Achselhöhlen und der Oberkörperseiten, tritt meist ab einem Alter von etwa 5–7 Jahre auf. Lisch-Knötchen treten oft erst während oder nach der Pubertät auf. Histologisch ist das Verhältnis von Melanozyten zu Keratinozyten, das bei der Neurofibromatose Typ 1 bereits in der nicht betroffenen Haut verschoben ist, im Bereich von Café-au-Lait-Flecken und Freckling weiter erhöht. Bei den im Bereich der Iris des Auges auftretenden Lisch-Knötchen handelt es sich histologisch um kleine pigmentierte Hamartome.
Veränderungen des Knochens und der Blutgefäße
NF1 kann schwerwiegende Auswirkungen auf die Knochenbildung haben. Einige der Störungen sind bei den Betroffenen bereits bei der Geburt vorhanden. Dazu gehören die Dysplasie des Keilbeinflügels, bei der die Augenhöhle deformiert ist. Eine andere angeborene Knochenstörung betrifft die langen Röhrenknochen der Extremitäten, welche verformt sein können oder zu Brüchen neigen und Pseudogelenke bilden können. Im Bereich der Blutgefäße kann eine fibromuskuläre Dysplasie auftreten, die insbesondere die Nierenarterien betreffen kann.
Im Altersbereich bis zu 12 Jahren kann es zu Verformungen der Wirbelkörper kommen, die zu einer schweren Skoliose führen können. Eine frühzeitige und regelmäßige Vorstellung bei einem mit NF1 vertrauten Spezialisten ist daher ratsam.
Klinische Manifestationen
Haut

Café-au-lait-Flecken und Farbveränderungen der Achsel sind auffällige Hauterscheinungen. In mehr als 95 Prozent der Fälle finden sich Café-au-lait-Flecken bei Patienten mit der Neurofibromatose Recklinghausen.[11] Etwa 80 Prozent weisen mehr als sechs große hyperpigmentierte Areale auf. Allerdings kommen Café-au-lait-Flecken auch bei etwa zehn Prozent der nicht betroffenen Bevölkerung vor. Es handelt sich bei dieser Veränderung um große (bis zu mehreren Zentimetern), scharf und unregelmäßig begrenzte hell- bis dunkelbraune Flecken, die oft schon bei Geburt oder in der Kindheit auffallen. Sie sind am Körper ohne erkennbare Ordnung verteilt. Es liegt eine Vermehrung von Melanozyten vor.
Das sogenannte Freckling (engl. freckle = Sommersprossen, Tüpfel, Sprenkel) ist eine sommersprossenähnliche Verfärbung an Körperstellen, die normalerweise keiner Sonnenbestrahlung ausgesetzt sind. Am auffälligsten sind diese Veränderungen in der Achselhöhle und Leistenregion. Dies wird ab einem Alter von etwa 10 Jahren beobachtet. Da in etwa 90 Prozent das Freckling bei Patienten mit Neurofibromatose Typ 1 auftritt, ist es eine diagnostisch wegweisende Erscheinung. Daneben werden auch diffuse Farbveränderungen des Rumpfes (Lentiginose) beschrieben, die ebenso gehäuft im Bereich der Axillen auftreten.
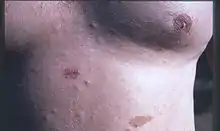
Neurofibrome sind Tumoren des peripheren Nervensystems, die sich vor allem im Bereich der Haut bemerkbar machen. Sie treten typischerweise kutan (Kutis = Gewebe der Haut), subkutan (Subkutis = Unterhautzellgewebe) oder als plexiforme Neurofibrome auf. Kutane Neurofibrome bilden sich typischerweise ab Beginn der Pubertät. Plexiforme Neurofibrome sind oft angeboren. Die Haut der Patienten kann im Lauf der Zeit mit bis zu 10.000 Tumoren unterschiedlicher Größe bedeckt sein. Sie variieren im Durchmesser von wenigen Millimetern bis zu mehreren Zentimetern je Läsion. Die Neurofibrome können unter der Oberfläche liegen und dann als hügelige Oberflächenstruktur der Haut auffallen. Andere können halbkugelig auf der Haut aufsitzen oder in Form eines Sackes anhaften. Auffällig ist, dass sie bei Druck in die Tiefe ausweichen, was als Knopflochphänomen oder Klingelknopfphänomen bezeichnet wird. Mit diesem einfachen Versuch kann man sehr leicht ein Neurofibrom von einem Lipom unterscheiden. Die Tumoren sind normalerweise hautfarben, können aber auch rötlich, bläulich oder violett erscheinen. Die kutanen Neurofibrome weisen eine weiche, homogene Konsistenz auf. Da die NF1 progredient ist, wächst die Anzahl der Neurofibrome ein Leben lang.
Die tiefer gelegenen subkutanen Neurofibrome sind derbe Verdickungen, die von den peripheren Nerven ausgehen. Da die Wucherungen auch auf die Nerven selbst drücken, führen sie häufig zu Schmerzen und Empfindungsveränderungen.
Die plexiformen Tumoren sind nicht selten im Gesicht, im Nacken, an der Hüfte und am Unterschenkel lokalisiert. Sie erreichen teilweise eine enorme Größe und zeigen den ungewöhnlichen Tastbefund multipler strangförmiger Gewächse („Sack voll Würmer“).
Nervensystem
ZNS-Tumoren (zum Beispiel das Pilozytische Astrozytom bei NF 1 und das Schwannom bei NF 2) und neurologische Symptome treten als ernstzunehmende Probleme der Neurofibromatose auf. Vor allem Tumoren der Hirnnerven können chirurgische Interventionen notwendig machen. Akustikus- und Trigeminus-Neurinome, die vorwiegend bei NF 2 auftreten, verursachen besonders Hörverlust, aber auch Schmerzen. Ein Foramen-Jugulare-Syndrom und Hypoglossus-Tumoren bewirken entsprechende Symptome, ein Optikus-Gliom kann eine einseitige Blindheit und Tumoren der Spinalwurzeln können Lähmungen und Schmerzen verursachen. Darüber hinaus werden verschiedene neurologische Symptome beschrieben: Betroffene Kinder haben oft einen niedrigen IQ, eine ADS oder ADHS, Probleme mit der Selbstorganisation, Motorik-Schwächen sowohl fein- als auch grobmotorisch, dadurch Schulschwierigkeiten, eher selten eine Epilepsie und bei Hypothalamus-Hamartomen eine Pubertas Präcox. Bei Auftreten von epileptischen Anfällen bei Patienten mit einer Neurofibromatose, kann dies als Zeichen dafür gelten, dass sich ein Hirntumor entwickelt.
Augen
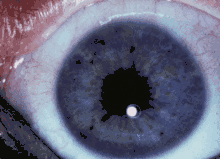
Die Lisch-Knötchen der Augen gelten als ein sehr hilfreiches diagnostisches Kriterium, da sie sich bei nahezu allen Patienten mit Neurofibromatose Typ 1 über 20 Jahren finden. Dabei handelt es sich um kleine, rundliche, scharf begrenzte und leicht erhabene Veränderungen in der Regenbogenhaut. Sie haben einen hellen, gelblich bis bräunlichen Farbton. Die Anzahl dieser Veränderungen nimmt mit dem Alter der Patienten zu. Diese von Melanozyten abstammenden gutartigen Gewebsveränderungen (Hamartome) der Iris wurden bereits 1918 von Waardenburg beschrieben.[13] Ihre Bedeutung für die Diagnose der Neurofibromatose wurde 1937 von Karl Lisch entdeckt. 1981 wurde durch die Arbeiten von Vincent M. Riccardi[14] und 1991 durch eine Studie von Marie Louise Lubs[15] der außerordentlich große Wert der Lisch-Knötchen für die Differentialdiagnose der Neurofibromatose Typ 1 herausgestellt.
Knochen
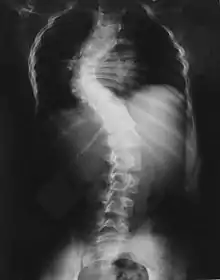
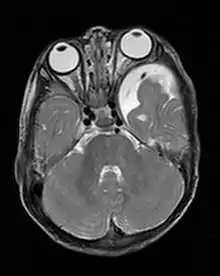
Skelettveränderungen treten bei einem Drittel der Patienten auf und bringen die Neurofibromatose-Patienten zum Orthopäden.
Sehr häufig finden sich Wirbelsäulenveränderungen von einfacher über kurzbogiger und knickförmiger Skoliose bis extrem ausgeprägten Kyphoskoliosen aufgrund von Fehlentwicklung der Wirbelkörper.[16]
Knochenzysten, Hypertrophien, pathologische Frakturen (Knochenbruch aufgrund einer Erkrankung des Knochens) und habituelle Luxationen (immer wiederkehrende Gelenk-Auskugelung) machen chirurgische Eingriffe notwendig. Minderwuchs und Vergrößerung (Megalenzephalie) oder Asymmetrie des Kopfes stellen für die Patienten belastende Symptome dar. Defekte der Orbitahinterwand verursachen manchmal einen pulsierenden Exophthalmus und täuschen so einen Tumor in der Augenhöhle vor.
Weitere charakteristische Veränderung ist die Verbiegung eines langen Röhrenknochens mit Frakturneigung und konsekutiver Pseudarthrose, am häufigsten in der Tibia (Kongenitale Tibiapseudarthrose).
Diagnose
Als Kardinalsymptome oder Kernsymptome bezeichnet man Merkmale, durch deren gemeinsames Auftreten eine Krankheit definiert ist. Bei Neurofibromatose Typ 1 werden folgende zwei Kardinalsymptome beschrieben. Mehr als 95 Prozent der Patienten mit einer gesicherten Neurofibromatose Typ 1 haben mehr als fünf Café-au-lait-Flecken, und bei mehr als 90 Prozent der Patienten findet man kutane Tumoren.
Als klinisches Spektrum bezeichnet man alle Symptome, die ein Patient mit einer bestimmten Erkrankung bekommen kann und deren Entstehung in einen kausalen Zusammenhang mit der Erkrankung gebracht wird, also nicht bloß zufällig ist. Bei den meisten Autoren gelten folgende Symptome als obligatorisches klinisches Spektrum der Neurofibromatose Typ 1: Café-au-lait-Flecken und kutane Neurofibrome zählen dazu. Der Nachweis von Lisch-Noduli gelingt je nach Studie bei 90 bis 100 Prozent der Patienten. Bei ca. 80 Prozent der Patienten findet sich eine sommersprossenartige Pigmentierung der Achselhöhle. Bei 20 Prozent der Patienten findet man große plexiforme Tumoren. Alle anderen Tumoren (spinale und periphere Neurofibrome, Schwannome der peripheren Nerven et cetera) finden sich bei weniger als fünf Prozent der Patienten. Etwa ein Drittel der Patienten hat darüber hinaus unspezifische Symptome wie Schulprobleme (30 Prozent), Minderwuchs (15 Prozent), Macrozephalie (25 Prozent) und Skoliosen (30 Prozent). Pseudoarthrosen und Epilepsien treten bei weniger als fünf Prozent der Patienten auf. Ein Teil der Patienten entwickelt ein Phäochromozytom.
Zu den diagnostischen Kriterien zählen die Symptome, die der allergrößte Teil der Patienten im Laufe der Erkrankung bekommt. Die folgende Tabelle gibt die diagnostischen Kriterien für die Neurofibromatose Typ 1 gemäß der NIH Consensus Development Conference von 1987 an:[17]
| Diagnostische Kriterien (zwei oder mehrere zutreffende Kriterien) | |
| 1) | Sechs Café-au-lait-Flecken (vor Pubertät größer als 5 mm, danach größer als 15 mm) |
| 2) | Axilläre oder inguinale Pigmentierung |
| 3) | Zwei oder mehr Neurofibrome oder ein plexiformes Neurofibrom |
| 4) | Ein Verwandter ersten Grades mit Neurofibromatose Typ 1 |
| 5) | Zwei oder mehr Lisch-Knötchen |
| 6) | Knochenläsionen |
Behandlung
Da es sich bei der Neurofibromatose Typ 1 um eine genetisch bedingte Erkrankung handelt, ist eine Therapie, die auf Heilung der zugrunde liegenden Störung abzielt, derzeit nicht möglich. Die einzige Behandlungsmöglichkeit besteht daher in der operativen Entfernung der Neurofibrome und Tumoren oder ausnahmsweise in deren Bestrahlung. Dabei sollte man allerdings sehr zurückhaltend vorgehen, denn die Operation eines Neurofibroms kann den Funktionsausfall des betreffenden Nervs mit bleibenden Lähmungen zur Folge haben. Bestrahlung kann ein vermehrtes Wachstum der Tumoren auslösen. Tumoren des zentralen Nervensystems können derart lokalisiert sein, dass ein operatives Vorgehen ohne Veränderungen an gesundem Gewebe nicht möglich ist. Es besteht außerdem die Möglichkeit, dass Operation und Bestrahlung ein Wachstum der Tumoren begünstigen können. Daher wird eine sehr genaue Risiko-Nutzen-Abwägung verlangt. Es werden üblicherweise nur solche Veränderungen entfernt, die das Risiko einer bösartigen Entwicklung besitzen. Hierbei kann es hilfreich sein einen PET-Scan durchzuführen.[18] Die Diagnose eines malignen peripheren Nervenscheidentumors sollte in Betracht gezogen werden, wenn ein Patient / eine Patientin Schmerzen entwickelt, die nicht erklärt werden können, die Größe eines Neurofibroms schnell zunimmt und / oder sich dessen Beschaffenheit ändert.[19] Auch eine schwere neurologische oder orthopädische Symptomatik, gravierende kosmetische Probleme sowie eine drohende Erblindung stellen Gründe für eine Operation dar.
Prognose
Aufgrund des in dem Abschnitt Pathogenese und Molekularbiologie beschriebenen Mechanismus entwickeln sich die meisten Symptome der Erkrankung erst im Laufe der Zeit. In diesem Sinne besteht auch eine Progredienz, wobei manche Tumoren, wie beispielsweise das Optikusgliom, auch über lange Zeit unverändert bleiben können. Mit der Vielfalt der genetischen Befunde gehen auch unterschiedliche Symptome und Verläufe der Erkrankung einher. Die Lebenserwartung der Patienten ist in vielen Fällen normal. Beim Auftreten maligner Tumoren wie maligner peripherer Nervenscheidentumoren oder Glioblastome ist die Lebenserwartung jedoch deutlich verringert.[20] Wegen des autosomal dominanten Erbganges ist wie bei anderen Erbkrankheiten eine genetische Beratung bei bestehendem Kinderwunsch sinnvoll.
Einzelnachweise
- M. Deckert, G. Reifenberger, U.-N. Riede, W. Schlote, D. R. Thal, O. D. Wiestler: Nervensystem. In: Ursus-Nikolaus Riede, Martin Werner, Hans-Eckart Schäfer (Hrsg.): Allgemeine und Spezielle Pathologie. 5. Auflage. Stuttgart, 2004, S. 1104.
- Bruce Ponder: Neurofibromatosis gene cloned. In: Nature. Band 346, 1990, S. 703. PMID 2117711
- David Viskochil: Deletions and a Translocation Interrupt a Cloned Gene at the Neurofibromatosis Type 1 Locus. In: Cell. Band 62, 1990, S. 187–192. PMID 1694727
- Margaret R. Wallace: Type 1 Neurofibromatosis Gene, Identification of a Large Transcript Disrupted in Three NF1 Patients. In: Science. Band 249, 1990, S. 181–186. PMID 2134734
- Toru Nishi: Differential expression of two types of the neurofibromatosis type 1 (NF1) gene transcripts related to neuronal differentiation. In: Oncogene. Band 6, 1991, S. 1555–1599. PMID 1923522
- Gangfeng Xu: The Neurofibromatosis Type 1 Gene Encodes a Protein Related to GAP. In: Cell. Band 62, 1990, S. 599–608. PMID 2116237
- Roymarie Ballester: The NF1 Locus Encodes a Protein Functionally Related to Mammalian GAP and Yeast IRA Proteins. In: Cell. Band 63, 1990, S. 851–859. PMID 2121371
- George A. Martin: The GAP-related Domain of the Neurofibromatosis Type 1 Gene Product interacts with ras p21. In: Cell. Band 63, 1990, S. 843. PMID 2121370
- Gangfeng Xu: The Catalytic Domain of the Neurofibromatosis Type 1 Gene Product Stimulates ras GTPase and Complements ira Mutants of S. cerevisiae. In: Cell. Band 63, 1990, S. 835–841. PMID 2121369
- von Deimling & Perry: Neurofibromatosis type 1. In: WHO classification of central nervous system tumors. Lyon, IAR Press, 2007.
- Anthony Killen, Emanuel Rubin, David Strayer: Developmental and Genetic Diseases. In: Raphael Rubin, David Strayer (Hrsg.): Rubin's Pathology. 5. Auflage. Philadelphia, 2008, S. 201.
- Manfred Abel: Notfallmedizinsiche und anästhesiologische Aspekte der Neurofibromatose im Kindesalter. In: Anästhesie Intensivtherapie Notfallmedizin. Band 20, Nr. 2, 1985, S. 76–78.
- P. J. Waardenburg: Heterochrome en melanosis. In: Nederlands tijdschrift voor geneeskunde. Band 62, 1918, S. 1453–1455.
- V. M. Riccardi: Neurofibromatosis: an overview and new directions in clinical investigations. In: Advances in neurology. Band 29, 1981, S. 1–9, ISSN 0091-3952. PMID 6798831.
- Marie-Louise Lubs: Lisch-Nodules in Neurofibromatosis Type I. In: The New England journal of medicine. Band 324, 1991, S. 1264. PMID 1901624
- F. Hefti: Kinderorthopädie in der Praxis. Springer 1998, ISBN 3-540-61480-X.
- Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. In: Archives of Neurology. Band 45, Nummer 5, Mai 1988, S. 575–578, ISSN 0003-9942. PMID 3128965.
- T. Derlin, J. Salamon, P. Bannas, J. D. Busch, J. Herrmann: Intratumorale Heterogenität der Traceraufnahme in der F-18 FDG PET/CT als Charakteristikum maligner peripherer Nervenscheidentumore bei Patienten mit Neurofibromatose Typ 1. In: RöFo - Fortschritte auf dem Gebiet der Röntgenstrahlen und der bildgebenden Verfahren. Band 185, S 1, April 2013, ISSN 1438-9029, S. VO310_2, doi:10.1055/s-0033-1346408 (thieme-connect.de [abgerufen am 30. Juli 2020]).
- Deutscher Ärzteverlag GmbH, Redaktion Deutsches Ärzteblatt: Tumoren peripherer Nerven: Ergänzung sinnvoll? 8. November 2002, abgerufen am 30. Juli 2020.
- Neurofibromatosis Fact Sheet. (Memento des Originals vom 19. November 2016 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. National Institute of Neurological Disorders and Stroke, abgerufen am 25. März 2012.
Literatur
Bücher
- Klaus Poeck, Werner Hacke: Neurologie. 11. Auflage. Springer, Berlin 2001, ISBN 3-540-41345-6.
- Peter Fritsch: Dermatologie und Venerologie. 2. Auflage. Springer, Berlin 2003, ISBN 3-540-00332-0.
- Lewis P. Rowland: Merrits Textbook of Neurology. Williams & Wilkins, Baltimore 1995, ISBN 0-683-07400-8.
- V. M. Riccardi (Hrsg.): Neurofibromatosis, phenotype, natural history and pathogenesis. Johns Hopkins University Press, Baltimore 1986, 1992, ISBN 0-8018-4348-0.
- Raymond D. Adams: Neurocutaneous Diseases. In: T. B. Fitzpatrick, A. Z. Eisen, K. Wolff (Hrsg.): Dermatology in General Medicine. 2 Bände. McGraw-Hill, New York 1987, 1993, 2003, ISBN 0-07-138076-0 (Set)
Artikel in Zeitschriften
- B. Castle: Evaluation of genotype-phenotype correlations in neurofibromatosis type 1. In: Journal of Medical Genetics. BMJ Publishing Group, London 40.2003,10 (Oct), 109. PMID 14569132 (Frei zugängliche Übersichtsarbeit)