AL-Amyloidose
Bei der AL-Amyloidose (auch Leichtketten-Amyloidose) handelt es sich um eine spezielle Form des hämatologischen Krankheitsbildes Amyloidose, wobei die hier beschriebene Unterform die häufigste Ausprägung der systemischen Amyloidosen darstellt und synonym auch als primäre Amyloidose bezeichnet wird. Sie ist abzugrenzen von der Leichtketten-Speicherkrankheit (light-chain deposition disease). Das Akronym AL steht für „Amyloid, bestehend aus Leichtketten“. Diese Leichtketten, die in ihrer freien Form von einem malignen Plasmazell-Klon gebildet werden, sind ursächlich für die Erkrankung. Freie Leichtketten finden sich allerdings auch in geringer Konzentration als natürliches Protein im Serum gesunder Patienten. Übergeordnet kann die AL-Amyloidose als Plasmazelldyskrasie, lymphoplasmozytische Erkrankung oder monoklonale Gammopathie bezeichnet werden. In den meisten Fällen tritt die AL-Amyloidose isoliert auf, steht aber auch bei 10–15 % der Fälle in Zusammenhang mit einem Multiplen Myelom und anderen monoklonalen Gammopathien.
| Klassifikation nach ICD-10 | |
|---|---|
| E85 | Amyloidose |
| ICD-10 online (WHO-Version 2019) | |
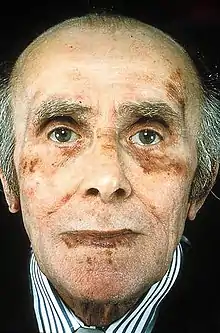
Verbreitung
Die AL-Amyloidose zählt zu den sogenannten seltenen Krankheiten. Für den deutschsprachigen Raum sind keine exakten Daten bekannt, da es für diese Erkrankung kein entsprechendes Register gibt. Es ist allerdings davon auszugehen, dass die Inzidenz bei etwa 5–13 Personen pro 1.000.000 Einwohner und Jahr liegt, wie sie auch bereits für die nordamerikanische Bevölkerung und in Großbritannien beschrieben wurde.[2][3][4] Männer sind dabei etwas häufiger betroffen als Frauen, wobei die Wahrscheinlichkeit einer Erkrankung mit zunehmendem Alter, ähnlich wie bei der monoklonalen Gammopathie unklarer Signifikanz (MGUS), ansteigt.[5][6][7] Das mediane Alter zum Zeitpunkt der Diagnose liegt bei 63–64 Jahren.[8]
Ursache
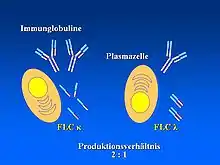
Ursächlich für die Erkrankung sind monoklonale freie Leichtketten (vom Typ Lambda (λ) oder Kappa (κ)) oder deren Fragmente, die von einer Form bösartig veränderter B-Zellen (den sogenannten monoklonalen Plasmazellen) produziert und ausgeschieden werden. Durch Fehlfaltung neigen diese zur Aggregation. Diese Aggregate bilden bei der AL-Amyloidose Fibrillen, die sich meist extrazellulär in fast allen Organen ablagern können.[9][10] Dadurch wird deren Funktion zunehmend beeinträchtigt, was zu dem typischen Erscheinungsbild der Krankheit führt (siehe Abschnitt Klinische Erscheinungen). Die Aminosäure-Sequenz und die dreidimensionale Struktur der Leichtketten kann bei betroffenen Patienten beeinträchtigt oder sogar verkürzt sein. Infolgedessen kann es in der dreidimensionalen Struktur zur Ausbildung eines β-Faltblatts und nicht zu der üblichen α-Helix kommen.[11] Diese strukturelle Abnormalität begünstigt die Aggregation und ist von diagnostischer Bedeutung beim Nachweis fehlgefalteter freier Leichtketten mittels Kongorot (siehe Abschnitt Untersuchungsmethoden).[12][13]
Betrachtet man die Bildung der Fibrillen im Detail, erweist sich der Prozess der Aggregation als hochkomplex, wobei die intrazelluläre Protein-Homöostase (Proteostase), extrazelluläre Chaperone sowie Matrix-Komponenten, Metall-Ionen, Scherkräfte, eine eingeschränkte Proteolyse und Zellinteraktionen eine Rolle spielen können.[9][14][15][16] Bei der Mehrzahl der betroffenen Patienten (80 %) ist eine monoklonale freie Leichtkette vom Typ λ ursächlich für die Erkrankung. Zytogenetisch ist bei > 50 % aller Patienten eine Translokation der Chromosomen 11 und 14 (t(11;14)) nachweisbar.[17] Weitere bekannte genetische Aberrationen sind die Translokation t(4;14), Zugewinn 1q21 und Deletion 17p13.
Krankheitsentstehung
Zytogenetische Veränderungen sind, wie im vorherigen Abschnitt bereits beschrieben, bekannt und gelten als Initiatoren der Krankheitsentstehung. Zudem konnte gezeigt werden, dass die Produktion monoklonaler freier Leichtketten bereits viele Jahre auftritt bevor es zur anschließenden Ablagerung kommt und sich Patienten mit Symptomen beim Arzt vorstellen.[18][19] Eine Abgrenzung von maligner zu nicht-maligner Erkrankung wird dadurch erschwert und ist nicht mehr mit der klassischen Ansicht der Signifikanz monoklonaler Gammopathien zu vereinbaren. Sobald eine monoklonale Gammopathie unklarer Signifikanz diagnostiziert wurde, bei der eine erhöhte Konzentration einer freien Leichtkette vorliegt, wird eine regelmäßige, engmaschige Untersuchung der betroffenen Patienten empfohlen, da das Risiko zur Entwicklung einer AL-Amyloidose erhöht ist.[20] Das "Screening" anhand kardialer und renaler Biomarker, dient hierbei als diagnostisches Mittel der Wahl (siehe auch Abschnitt Vorbeugung und Früherkennung).[21][22]
Klinische Erscheinungen
Ähnlich wie bei anderen monoklonalen Gammopathien sind die vom Patienten wahrgenommenen Symptome der AL-Amyloidose typisch aber auch sehr unspezifisch. Viele Patienten klagen über Müdigkeit und Schwächegefühl sowie Knochenschmerzen. Auch Ödeme, eine Parästhesie und Gewichtsverlust können unter Umständen bei betroffenen Patienten zum Zeitpunkt der Erstvorstellung beobachtet werden.[23] Eine solche klinische Erscheinung kann zur Einleitung weiterer Untersuchungen dienen. Hier können dann auch die für die AL-Amyloidose typischen Ablagerungen in Organen – insbesondere in der Niere und im Herzen – sowie eine Polyneuropathie als Ursache der Parästhesie, bestätigt oder widerlegt werden. Ablagerungen in Niere und/oder Herzen können sich jeweils z. B. in einem nephrotischen Syndrom bzw. einer nicht-ischämischen Kardiomyopathie mit Hypertrophie in der Echokardiografie äußern. Liegt eine Beeinträchtigung der Nierenfunktion vor, kann die AL-Amyloidose auch zur Gruppe der monoklonalen Gammopathien renaler Signifikanz gezählt werden. Neben den zuvor genannten Leitsymptomen können eine Hepatomegalie oder erhöhte Werte der Alkalischen Phosphatase ohne morphologische Abnormalitäten sowie eine Proteinurie vorliegen, welche häufig fälschlicherweise dem abzugrenzenden Multiplen Myelom mit Cast-Nephropathie zugerechnet wird.[23]
Prognose
Laut einer schwedischen Studie liegt das mediane Überleben betroffener Patienten bei etwa 3 Jahren.[24] Bis zu 30 % aller Patienten sterben sogar innerhalb des ersten Jahres nach Diagnosestellung.[25][26] Wenn die Erkrankung nicht therapiert wird, versterben ungefähr 80 % der Patienten bereits innerhalb der ersten zwei Jahre nach Diagnosestellung, wobei dies in den meisten Fällen auf eine kardiale Dysfunktion zurückzuführen ist.[26][27][28] Je früher die Diagnose erfolgt desto besser ist auch die Prognose für betroffene Patienten.
Bei Diagnose und Einleitung einer Therapie vor Ausbildung einer fortgeschrittenen Kardiomyopathie, ist ein hämatologisches Ansprechen sowie ein Ansprechen auf Organebene möglich, was sich auch in einem längeren Überleben der Patienten widerspiegelt.*[23] Durch moderne Therapeutika kann die Erkrankung bei rechtzeitiger Therapieeinleitung häufig gut kontrolliert werden.
Untersuchungsmethoden
Eine umfassende diagnostische Abklärung sollte in Anbetracht der Schwere der Erkrankung bei einem dringenden Verdacht durchgeführt werden. Aufgrund der unspezifischen Symptomatik, die häufig bei älteren Patienten vorliegt, fällt eine Verdachtsäußerung deshalb unter Umständen erst spät. Ähnlich wie bei anderen monoklonalen Gammopathien enthält die Diagnostik neben der grundlegenden Anamnese mit körperlicher Untersuchung eine Reihe verschiedener Laborparameter, bildgebende Methoden sowie histologische Untersuchungen. Empfehlungen finden sich in Publikationen verschiedener nationaler sowie internationaler Forschungsgesellschaften. So wird beispielsweise bei allen Patienten, die bereits mit einer monoklonalen Gammopathie unklarer Signifikanz diagnostiziert sind, auf das potenzielle Vorliegen einer AL-Amyloidose hingewiesen.[23] Das aktuell anerkannte diagnostische Spektrum setzt sich wie folgt zusammen:[22]
Laborparameter
Zentrale Laborparameter zum Nachweis der monoklonalen freien Leichtketten bei Verdacht auf eine AL-Amyloidose sind:[29][30][8]
- Immunfixationselektrophorese einer Serum- sowie einer 24h-Urin-Probe
- quantitative Bestimmung der freien Leichtketten Kappa- und Lambda im Serum (inkl. Berechnung des Quotienten (bzw. Ratio, rFLC) als auch der Differenz (dFLC) aus involvierter freier Leichtkette (iFLC) und nicht-involvierter freier Leichtkette (uFLC))
Bei negativer Serum- als auch Urin-Immunfixationselektrophorese sowie normaler rFLC ist eine AL-Amyloidose unwahrscheinlich und weitere Untersuchungen nur bei bestehendem dringenden Verdacht empfohlen.[23]
Weitere mögliche Untersuchungen sind:
- Blutbild
- Differentialblutbild
- 24h-Urin-Probe zur Quantifizierung der Gesamteiweiß- und Albumin-Ausscheidung sowie zur Diagnose einer Bence-Jones-Proteinurie
- Serumproteinelektrophorese bzw. Urinproteinelektrophorese (einer 24h-Urin-Probe)
- Bestimmung der/des
Bildgebende Diagnostik
- Projektionsradiographie oder sensitivere, wenn auch strahlenintensivere Low-Dose-CT zur Diagnostik von Osteolysen und zur Stabilitätsbeurteilung
- CT des Thorax
- Spirometrie bei Verdacht auf pulmonale Amyloidose
- MRT bei Verdacht auf Weichteil-Manifestationen
- Transthorakale Echokardiographie, EKG
- ggf. ein Kardio-MRT sowie ein 24h-EKG (bei Rhythmusstörungen und eingeschränkter Herzfrequenzvariabilität)
- Sonografie des Abdomen (Bestimmung von Leber- und Milz-Größe, Restharnmessung)
- Neurologische/elektrophysiologische Untersuchung
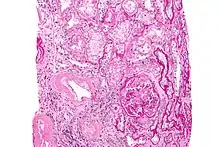
Zytologische/histologische Untersuchungen
- Knochenmarkpunktion
- Biopsie zur Bestimmung des prozentualen Anteils klonaler Plasmazellen und Amyloid-Nachweis
- Multiparametrische Immunphänotypisierung des Knochenmarks zum Ausschluss einer zugrunde liegenden Lymphomerkrankung
- Optional: FISH-Untersuchung auf Translokationen t(11;14) sowie t(4;14), Zugewinn 1q21 und Deletion 17p13.
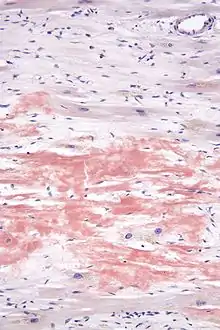
- Gewebebiopsie mit Kongorot-Färbung von subkutanem Fettgewebe, dem Rektum (tief) oder Knochenmark
Der Kongorotfärbung einer Gewebebiopsie ist hinzuzufügen, dass auch häufig auf Proben der vergangenen 1–2 Jahre zur nachträglichen Anfärbung zurückgegriffen werden kann, soweit solche Proben bereits vorliegen. Zur eindeutigen Diagnose ist eine Biopsie integraler Bestandteil. Bei einem positiven Befund (polarisationsoptische Doppelbrechung in grüner oder rot-gelblicher Farbe), muss das Amyloid (immunhistologisch oder ggf. mittels Massenspektrometrie) typisiert werden um die zugrundeliegende Amyloidose-Form zu bestätigen, da die Kongorotfärbung nicht spezifisch für die AL-Amyloidose ist.[31][32][33]
Therapie
Die effektivste Therapiemöglichkeit besteht in der Kombination aus autologer Stammzelltransplantation (soweit der Allgemeinzustand des Patienten diesen Eingriff erlaubt) und gleichzeitiger Chemotherapie, vergleichbar mit der, die auch beim Multiplen Myelom zum Einsatz kommt.[21] Die Gabe von Melphalan und Dexamethason hat sich als effektiv für Patienten erwiesen, die nicht für eine autologe Stammzelltransplantation geeignet sind. Im Gegensatz dazu erwies sich eine Kombination aus Dexamethason und Bortezomib als besonders geeignet für eine gewisse Gruppe anderer Patienten.[21] Seit Juli 2021 ist auch die Kombination des Antikörpers Daratumumab mit Cyclophosphamid, Bortezomib und Dexamethason zur Behandlung der AL-Amyloidose zugelassen.[34] Therapieentscheidungen inkl. der am besten geeigneten Therapeutika-Kombination müssen immer für jeden Patienten individuell vom behandelnden Arzt getroffen werden.
Vorbeugung und Früherkennung
Aufgrund der Komplexität der Krankheitsentstehung und dem mangelhaften Verständnis darüber, sind keine allgemeingültigen Maßnahmen zur Vorbeugung der Krankheit bekannt. Bestimmte Umwelteinflüsse wie Übergewicht, Pestizide, Strahlung, Autoimmunerkrankungen sowie häufige Entzündungen und Infektionen gelten als Risikofaktoren der monoklonalen Gammopathie unklarer Signifikanz, die der AL-Amyloidose vorausgeht. Bei MGUS-Patienten konnte zudem ein stark abnormer Quotient der freien Leichtketten (Kappa/Lambda < 0,01 bzw. > 100) als Risikofaktor der Progression zu einer AL-Amyloidose (sowie dem Multiplen Myelom) identifiziert werden.[35][20]
Eine frühzeitige Diagnose der Erkrankung, bzw. des nicht-malignen Vorläufers MGUS, ist entscheidend für den Krankheitsverlauf und den Zustand des Patienten. Eine späte Diagnose der Erkrankung, welche aufgrund der oft fehlgedeuteten Symptome häufig ist, stellt den Grund für den hohen Anteil Betroffener (ca. 25 %) mit einem fortgeschrittenen Erkrankungsstadium dar. Die Ausbildung erster Fibrillen begünstigt die Ausbildung weiterer Ablagerungen, weswegen eine umgehende Therapieeinleitung notwendig ist um weitere negative Folgen zu unterbinden. Je stärker die Produktion pathogener freier Leichtketten unterbunden werden kann, desto positiver der zu erwartende Allgemeinzustand des Patienten. Selbst geringe Konzentrationen pathogener freier Leichtketten können noch für die Ausbildung weiterer Ablagerungen genügen. Amyloide Firbrillen gelten zwar als resistent gegen Abbau, scheinen allerdings bei gutem Therapieansprechen nach und nach vom Körper reabsorbiert zu werden.[21]
Da kardiologische Folgen der AL-Amyloidose als besonders schwerwiegend zu erachten sind und für Betroffene eine geringe Lebenserwartung bedeuten, empfehlen die Experten des Amyloidosis Research Consortium generell alle Patienten mit Herzinsuffizienz (mit konzentrischer Hypertrophie oder NYHA I-II oder mit beiden Befunden) auf das Vorliegen erhöhter freier Leichtketten untersucht werden.[36] Bei Vorliegen einer MGUS mit einem abnormalen Quotienten der freien Leichtketten, wird von Experten ein "Screening" dieser Patienten auf die Biomarker NT-proBNP (präsymptomatisch abnormal bei kardiologischen Komplikationen) und Albumin (präsymptomatisch abnormal bei renalen Komplikationen) empfohlen.[21] Einem schwerwiegenden Krankheitsverlauf kann dadurch unter Umständen vorgebeugt und somit eine bessere Lebensqualität erreicht werden.
Es konnte gezeigt werden, dass Patienten in Risikogruppen (Alter, MGUS-Befund liegt bereits vor, Herzinsuffizienz etc.) von einer regelmäßigen Untersuchung profitieren, da eine maligne Erkrankung frühzeitig erkannt und entsprechende Maßnahmen ergriffen werden können.[37]
Weblinks
- Onkopedia - Deutschsprachige Leitlinien zur AL-Amyloidose
- International Myeloma Foundation Deutschsprachiges Patientenhandbuch
- Wikilite: AL-Amyloidose - Englischsprachige Seite mit detaillierten Informationen zur AL-Amyloidose und monoklonalen Gammopathien im Allgemeinen
- Myelom Deutschland e. V. Weiterführende Informationen zur AL-Amyloidose für Patienten
- Arbeitsgemeinschaft Multiples Myelom Weiterführende Informationen zur AL-Amyloidose für Patienten
- Deutsche Krebshilfe Plasmozytom/Multiples Myelom. Antworten. Hilfen. Perspektiven - Blauer Ratgeber #22
Einzelnachweise
- RA KYle et al.: Primary systemic amyloidosis: clinical and laboratory features in 474 cases. In: Semin Hematol. 32, Nr. 1, Januar 1995, S. 45–59. PMID 7878478.
- MA Gertz et al.: The classification and typing of amyloid deposits. In: Am J Clin Pathol. 121, Nr. 6, Juni 2004, S. 787–9. doi:10.1309/TR4L-GLVR-JKAM-V5QT. PMID 15198347.
- J Bird et al.: Guidelines on the diagnosis and management of AL amyloidosis. In: Br J Hematol. 125, Nr. 6, 2004, S. 681–700. doi:10.1111/j.1365-2141.2004.04970.x. PMID 15180858.
- RA Kyle et al.: Incidence and natural history of primary systemic amyloidosis in Olmsted CountyIncidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. In: Blood. 79, Nr. 7, 1992, S. 1817–1822. PMID 1558973.
- UNC: AL Amyloidosis. UNC. Archiviert vom Original am 22. Dezember 2011. Abgerufen am 7. März 2018.
- Primary AL. Amyloidosis Foundation. Archiviert vom Original am 3. Oktober 2011. Abgerufen am 7. März 2018.
- RA Kyle et al.: Monoclonal gammopathy of undetermined significance. In: Br J Hematol. 134, Nr. 6, September 2006, S. 573–589. doi:10.1111/j.1365-2141.2006.06235.x. PMID 16938117.
- G Palladini et al.: New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. In: J Clin Oncol. 30, Nr. 36, 2012, S. 4541–4549. doi:10.1200/JCO.2011.37.7614. PMID 23091105.
- G Merlini et al.: Molecular mechanisms of amyloidosis. In: N Engl J Med. 349, Nr. 6, 2003, S. 583–596. doi:10.1056/NEJMra023144. PMID 12904524.
- G Merlini et al.: Amyloidosis: pathogenesis and new therapeutic options. In: J Clin Oncol. 29, Nr. 14, 2011, S. 1924–1933. doi:10.1200/JCO.2010.32.2271. PMID 21483018. PMC 3138545 (freier Volltext).
- A Bhat et al.: Current concepts on the immunopathology of amyloidosis. In: Clin Rev Allergy Immunol. 38, Nr. 2–3, 2010, S. 97–106. doi:10.1007/s12016-009-8163-9. PMID 19626465.
- MM Picken et al.: Amyloidosis: Where are we now and where are we heading?. In: Arch Pathol Lab Med. 134, Nr. 4, 2010, S. 545–551. doi:10.1043/1543-2165-134.4.545. PMID 20367306.
- MM Picken et al.: Amyloid detection and typing: summary of current practice and recommendations of the consensus group. In: Amyloid. 18 Suppl 1, 2011, S. 48–50. doi:10.3109/13506129.2011.574354017. PMID 20367306.
- F Chiti et al.: Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. In: Annu Rev Biochem. 86, 2017, S. 27–68. doi:10.1146/annurev-biochem-061516-045115. PMID 28498720.
- AR Wyatt et al.: Extracellular chaperones and proteostasis. In: Annu Rev Biochem. 82, 2013, S. 295–322. doi:10.1146/annurev-biochem-072711-163904. PMID 23350744.
- D Ami et al.: In situ characterization of protein aggregates in human tissues affected by light chain amyloidosis: a FTIR microspectroscopy study. In: Sci Rep. 6, 2016. doi:10.1038/srep29096. PMID 27373200. PMC 4931462 (freier Volltext).
- T Bochtler et al.: Evaluation of the serum-free light chain test in untreated patients with AL amyloidosis. In: Haematologica. 93, Nr. 3, 2008, S. 459–462. doi:10.3324/haematol.11687. PMID 18287131.
- BM Weiss et al.: A monoclonal gammopathy precedes multiple myeloma in most patients. In: Blood. 113, Nr. 22, 2009, S. 5418-22. doi:10.1182/blood-2008-12-195008. PMID 19234139. PMC 2689043 (freier Volltext).
- BM Weiss et al.: Increased serum free light chains precede the presentation of immunoglobulin light chain amyloidosis. In: J Clin Oncol. 32, Nr. 25, 2014, S. 2699-704. doi:10.1200/JCO.2013.50.0892. PMID 25024082. PMC 4145182 (freier Volltext).
- N van de Donk et al.: The clinical relevance and management of monoclonal gammopathy of undetermined significance and related disorders: recommendations from the European Myeloma Network. In: Haematologica. 99, Nr. 6, 21. März 2014, S. 984–96. doi:10.3324/haematol.2013.100552. PMID 23224402. PMC 4040895 (freier Volltext).
- G Merlini et al.: AL amyloidosis: from molecular mechanisms to targeted therapies. In: Hematology Am Soc Hematol Educ Program. Nr. 1, 8. Dezember 2017, S. 1–12. doi:10.1182/asheducation-2017.1.1. PMID 29222231.
- DGHO Leitlinie „AL-Amyloidose“
- MA Gertz et al.: Immunoglobulin light chain amyloidosis: 2016 update on diagnosis, prognosis, and treatment. In: Am J Hematol. 91, Nr. 9, 9. September 2016, S. 947-56. doi:10.1002/ajh.24433. PMID 27527836.
- K Heminki et al.: Incidence and survival in non-hereditary amyloidosis in Sweden. In: BMC Public Health. 12, 2012, S. 974. doi:10.1186/1471-2458-12-974. PMID 23148499. PMC 3503866 (freier Volltext).
- G Merlini et al.: Amyloidosis: is a cure possible?. In: Ann Oncol. 19 Suppl 4, 2009, S. iv63-6. doi:10.1093/annonc/mdn200. PMID 18519408.
- J Gillmore et al.: Guidelines on the diagnosis and investigation of AL amyloidosis. In: BJH. 168, Nr. 2, 2015, S. 207-18. doi:10.1111/bjh.13156. PMID 25312307.
- RL Comenzo et al.: Consensus guidelines for the conduct and reporting of clinical trials in systemic light-chain amyloidosis. In: BMC Public Health. 26, Nr. 11, 2012, S. 2317-25. doi:10.1038/leu.2012.100. PMID 22475872.
- S Kumar et al.: Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. In: J Clin Oncol. 30, Nr. 9, 2012, S. 989-95. doi:10.1200/JCO.2011.38.5724. PMID 22331953. PMC 3675680 (freier Volltext).
- A Dispenzieri et al.: International Myeloma Working Group guidelines for serum-free light chain analysis in multiple myeloma and related disorders. In: Leukemia. 23, Nr. 2, 20. November 2008, S. 215–224. doi:10.1038/leu.2008.307. PMID 19020545.
- JA Katzmann et al.: Screening Panels for Detection of Monoclonal Gammopathies. In: Clin Chem. 55, Nr. 8, 2009, S. 1517–1522. doi:10.1373/clinchem.2009.126664. PMID 19520758. PMC 3773468 (freier Volltext).
- HJ Lachmann et al.: Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. In: N Engl J Med. 346, Nr. 23, 2002, S. 1786–1791. doi:10.1056/NEJMoa013354. PMID 12050338.
- RL Comenzo et al.: Seeking confidence in the diagnosis of systemic AL(Ig light-chain) amyloidosis: patients can have both monoclonal gammopathies and hereditary amyloid proteins. In: Blood. 107, Nr. 2, 2006, S. 3489–3491. doi:10.1182/blood-2005-10-4148. PMID 16439680.
- SO Schönland et al.: Immunohistochemistry in the classification of systemic forms of amyloidosis: a systematic investigation of 117 patients. In: Blood. 119, Nr. 9, 2012, S. 488–493. doi:10.1182/blood-2011-06-358507. PMID 22106346.
- European Medicines Agency: EPAR Darzalex. 8. März 2021, abgerufen am 6. August 2021.
- RA Kyle et al.: Clinical course of light-chain smouldering multiple myeloma (idiopathic Bence Jones proteinuria): a retrospective cohort study. In: The Lancet Haematology. 1, Nr. 1, 1. Oktober 2014, S. e28-e36. doi:10.1016/S2352-3026(14)70001-8. PMID 25530988. PMC 4266993 (freier Volltext).
- MS Maurer et al.: Recommendations From the Amyloidosis Research Consortium Educational Roundtable at the American College of Cardiology Annual Meeting, April 1, 2016. In: Amyloid. 24(sup1), März 2017, S. 165–166. doi:10.1080/13506129.2017.1286582. PMID 28434366.
- RS Go et al.: Determining the clinical significance of monoclonal gammopathy of undetermined significance: a SEER-Medicare population analysis. In: Clin Lymphoma Myeloma Leuk. 15, Nr. 3, 2014, S. 177–186 e4. doi:10.1016/j.clml.2014.09.004. PMID 23224402. PMC 4344843 (freier Volltext).