Kernspinresonanzspektroskopie
Die Kernspinresonanzspektroskopie (NMR-Spektroskopie von englisch nuclear magnetic resonance) ist eine spektroskopische Methode zur Untersuchung der elektronischen Umgebung einzelner Atome und der Wechselwirkungen mit den Nachbaratomen. Dies ermöglicht die Aufklärung der Struktur und der Dynamik von Molekülen sowie Konzentrationsbestimmungen.

Die Methode beruht auf der magnetischen Kernresonanz, einer resonanten Wechselwirkung zwischen dem magnetischen Moment von Atomkernen der Probe, die sich in einem starken statischen Magnetfeld befindet, mit einem hochfrequenten magnetischen Wechselfeld. Es sind nur solche Isotope der Spektroskopie zugänglich, die im Grundzustand einen von Null verschiedenen Kernspin und damit ein magnetisches Moment besitzen, zum Beispiel 1H; 2D; 6Li; 10B; 11B; 13C; 15N; 17O; 19F; 31P und 43Ca.
Geschichte

Zur Vorgeschichte der Kernspinresonanzspektroskopie siehe Geschichte und Entwicklung der Kernspinresonanz.
Felix Bloch und Edward Mills Purcell wiesen erstmals 1946 Signale der magnetischen Kernresonanz nach, wofür sie 1952 den Nobelpreis erhielten. In ihrem Nobelpreisvortrag zeigten sie erste Spektren mit dem Nachweis der chemischen Verschiebung (am Beispiel des Ethanol), womit die eigentliche Kernspinspektroskopie begann. Sie entwickelte sich zu einer wichtigen Methode in der chemischen Strukturaufklärung. Zunächst wurde hauptsächlich die Continuous-Wave-(CW)-Methode benutzt, bei der durch Variation der Frequenz oder des Feldes die Resonanzen nacheinander angeregt wurden.[1] 1947 reichten Russell Varian und Felix Bloch ein Patent ein für das erste Kernspinresonanz-Spektrometer. Das erste kommerzielle Kernspinresonanz-Spektrometer baute 1952 die Firma Varian Associates in Palo Alto. Um 1955 baute die japanische Firma JEOL ebenfalls NMR-Spektrometer. Die US-amerikanische Biophysikerin Mildred Cohn setzte in den frühen 1960er Jahren die Kernspinresonanz-Spektroskopie zur Aufklärung metabolischer Prozesse auf molekularer Ebene ein.[2]

Da die CW-Technik durch ein schlechtes Signal-Rausch-Verhältnis gekennzeichnet war, entwickelte ab Mitte der 1960er Jahre Richard R. Ernst (Nobelpreis für Chemie 1991) bei der Firma Varian ein Puls-Fourier-Transformation-NMR-Spektrometer (FT-NMR), das eine wesentlich schnellere Aufnahme der Spektren ermöglichte. Bei gleicher Messzeit bedeutete das im Vergleich zu den CW-Spektrometern eine wesentliche Steigerung der Empfindlichkeit und damit des Signal-Rausch-Verhältnisses.[3] Bereits in den Jahren 1949 und 1950 waren von Hahn und Torrey die ersten Pulsverfahren untersucht worden.[4] Die ersten kommerziellen Kernspinresonanz-Impulsspektrometer wurden Mitte der 1960er Jahre von der deutschen Firma Bruker[5] (gegründet von Günther Laukien, einem der NMR-Pioniere in Deutschland) in Karlsruhe von einer Gruppe um Bertold Knüttel und Manfred Holz[6] gebaut. Es folgte die Einführung von Breitbandentkopplung und von Mehrpulsverfahren. Nach einer Idee von Jean Jeener wurden ab Anfang der 1970er Jahre Mehrpulsexperimente mit einer systematisch variierten Wartezeit zwischen zwei Pulsen entwickelt, die nach Fourier-Transformation über zwei Zeitbereiche zu zweidimensionalen Spektren führten.
Kurt Wüthrich und andere bauten diese 2D- und Multi-Dimensions-NMR zu einer bedeutenden Analysetechnik der Biochemie aus, insbesondere zur Strukturanalyse von Biopolymeren wie Proteinen. Wüthrich bekam für diese Arbeiten 2002 den Nobelpreis in Chemie.[7] Im Gegensatz zur Röntgenstrukturanalyse liefert die Kernspinresonanz-Spektroskopie Strukturen von Molekülen in Lösung. Von besonderer Bedeutung ist die Möglichkeit, detaillierte Informationen über die Moleküldynamik mit Hilfe von Relaxationsparametern zu gewinnen.
Physikalischer Hintergrund
Quantenmechanische Grundlagen
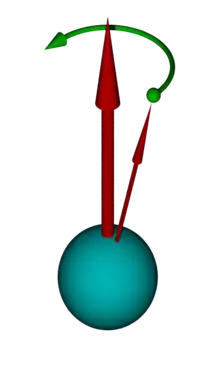
Teilchen und Atomkerne, die einen Kernspin besitzen, haben als rotierende Ladungsträger ein magnetisches Moment, das oft mit bezeichnet wird. Das magnetische Moment von Atomkernen kann in einem äußeren Magnetfeld nicht jede beliebige, sondern nur bestimmte, durch die Quantenmechanik beschriebene Orientierungen einnehmen. Die Zahl der möglichen Orientierungen wird durch die Kernspinquantenzahl bestimmt (siehe: Multiplizität). Zu jeder Kernspinquantenzahl existieren Orientierungen und jeder Orientierung ist eine magnetische Kernspinquantenzahl zugeordnet.





Beispiele:
- Wasserstoff-Kern mit Kernspin : zwei Orientierungen mit und
- Deuterium-Kern mit Kernspin : drei Orientierungen mit , und







Ohne ein äußeres Magnetfeld sind die mit gekennzeichneten Zustände energetisch gleich (siehe Entartung (Quantenmechanik)). In Anwesenheit eines äußeren Magnetfeldes entstehen Energiedifferenzen (Zeeman-Effekt). Kernresonanz-Phänomene beruhen auf der Anregung von Kernspin-Übergängen zwischen solchen -Zuständen. Die dazu benötigte Energie ist proportional zur Stärke des äußeren Magnetfeldes und zum gyromagnetischen Verhältnis des betrachteten Atomkerns:




Diese Energie wird durch Einstrahlen von resonanten elektromagnetischen Wellen eingebracht. Die Resonanzfrequenz wird in der NMR-Spektroskopie als Larmor-Frequenz bezeichnet und liegt im Radiowellen-Bereich. Gängige NMR-Spektrometer arbeiten bei Protonen-Resonanzfrequenzen zwischen 300 und 1000 MHz, was Feldstärken zwischen 7 und 24 Tesla erfordert.

Wenn alle 1H- oder 13C-Atome die exakt gleiche Larmor-Frequenz hätten, wäre die NMR-Methode zur Strukturaufklärung wenig interessant. Tatsächlich hängen aber die Resonanzfrequenzen von den individuellen, atomar aktiven Magnetfeldern ab. Diese lokalen Magnetfelder können in ihrer Stärke vom Hauptmagnetfeld abweichen, beispielsweise durch den Einfluss der elektronischen Umgebung eines Atomkerns oder durch magnetische Wechselwirkung zwischen benachbarten Atomkernen. Aufgrund dieser Eigenschaften wird die Kernresonanzspektroskopie zur Strukturaufklärung von Molekülen eingesetzt.
Messverfahren der Kernresonanzspektroskopie
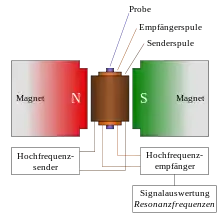
Zur Messung wird die Probe in ein homogenes magnetisches Feld gebracht, das sogenannte Hauptmagnetfeld. Die Probe wird von einer Induktionsspule umgeben, welche ein hochfrequentes elektromagnetisches Wechselfeld senkrecht zum Hauptmagnetfeld erzeugt. Dann variiert man die Stärke des Hauptmagnetfeldes, bis der Resonanzfall eintritt (Continuous-Wave-Verfahren, veraltet). Alternativ kann die magnetische Feldstärke konstant gehalten und die Frequenz des eingestrahlten Wechselfeldes variiert werden (Continuous-Field-Verfahren, veraltet). Wenn der Resonanzfall eintritt, die Probe also Energie aus dem Wechselfeld aufnimmt, verändert sich die Stromstärke, welche zum Aufbau des Wechselfeldes benötigt wird. Dies ist messbar.
Moderne Messverfahren strahlen nicht mehr kontinuierliche Wechselfelder in die Probe ein, sondern Radiowellen-Pulse. Ein kurzer Radiowellenpuls regt dabei ein Frequenzband an, dessen Frequenzbreite über die Fourier-Beziehung umgekehrt proportional zur Pulsdauer ist. Dadurch werden in der Probe alle Übergänge, die in dieses Frequenzband fallen, gleichzeitig angeregt. Bei korrekter Wahl von Pulsdauer und Pulsleistung kann die Magnetisierung der angeregten Kernspins in die Transversalebene senkrecht zum Hauptmagnetfeld gebracht werden. Nach Beendigung des Pulses oszilliert diese Transversalmagnetisierung für kurze Zeit senkrecht zum Hauptmagnetfeld. Dabei oszilliert jeder Kernspin mit seiner individuellen Larmor-Frequenz. Diese Summe dieser Oszillationen wird als elektrischer Strom über elektromagnetische Induktion mit der gleichen Induktionsspule detektiert, die zum Senden des Anregungspulses gedient hat. Das empfangene Signal wird digitalisiert und aufgezeichnet. Mit Hilfe der schnellen Fourier-Transformation ist es möglich, die individuellen Larmor-Frequenzen aus der Summe der Oszillationen zu extrahieren, um ein NMR-Spektrum zu erhalten. Darum tragen moderne NMR-Verfahren den Namen PFT-NMR für Pulsed Fourier Transform NMR Spectroscopy. Für dieses Verfahren bleibt das Hauptmagnetfeld statisch, üblicherweise wird es mit Hilfe von supraleitenden Elektromagneten erzeugt, die mit flüssigem Helium und Stickstoff gekühlt werden.
Ein spezielles Verfahren ist die NMR-CIDNP-Spektroskopie, bei der die Probe zur Erzeugung von Radikalen mit Licht bestrahlt oder beheizt wird.
Relaxation
Bei der PFT-NMR existieren drei Relaxationsprozesse, die einerseits die Leistungsfähigkeit der PFT-NMR einschränken, aber andererseits einzigartige Informationen zur Molekül-Dynamik und Material-Inhomogenitäten liefern können. Die -Relaxation ist der Prozess der Rückkehr der Kernspins vom angeregten Zustand zum thermischen Gleichgewicht unter Abgabe der bei der Anregung aufgenommenen Energie als Wärme. Wird zwischen zwei NMR-Experimenten (scans) nicht die vollständige -Relaxation abgewartet, so steht für jedes weitere nur eine gewisse, geringere Magnetisierung und damit Signalintensität zur Verfügung. Falls eine kurze Repetitionzeit gewünscht ist, muss der Anregungswinkel (auf den sog. Ernst-Winkel) verkleinert werden, um trotzdem ein möglichst starkes Signal zu erhalten. Die -Relaxation ist die Dephasierung der Transversalmagnetisierung aufgrund entropischer Effekte, die mit der magnetischen Dipol-Dipol-Wechselwirkung benachbarter Atomkerne zusammenhängt. Hier wird keine Energie abgegeben, da die Spins im angeregten Zustand verbleiben, aber die Transversalmagnetisierung läuft von einem Vektor zunächst zu einem Fächer und zuletzt zu einer Kreisfläche auseinander, so dass kein NMR-Signal mehr in der Detektionsspule induziert wird. Befinden sich zusätzlich Magnetfeldinhomogenitäten in der Probe, sei es durch Imperfektionen des Hauptmagnetfeldes oder durch Suszeptibilitätsunterschiede innerhalb der Probe, wird statt der -Relaxation die beschleunigte -Relaxation beobachtet.



Die -Relaxation bzw. -Relaxation beschränkt die Lebensdauer des NMR-Signals direkt nach der Anregung. Das NMR-Signal wird darum als gedämpfte Schwingung, als FID (free induction decay) gemessen. In der Praxis wird die -Relaxationszeit mit Hilfe der Spin-Echo-Methode gemessen.
Die -Relaxation limitiert, wie schnell NMR-Experimente hintereinander ausgeführt werden können. Die -Relaxation und die -Relaxation hängen stark von der Dichte und Viskosität/Rigidität der Probe ab.
Empfindlichkeit der NMR-Spektroskopie
Ein inhärentes Problem der NMR-Spektroskopie ist ihre vergleichsweise geringe Empfindlichkeit (schlechtes Signal-Rausch-Verhältnis). Dieses ist darauf zurückzuführen, dass die Energiedifferenzen der -Zustände klein und die Populationsunterschiede zwischen den Zuständen im thermischen Gleichgewicht sehr gering sind (Boltzmannverteilung).
Das Besetzungsverhältnis der beiden beteiligten Energiezustände kann durch deren Energiedifferenz im Verhältnis zur thermischen Energie bei gegebener Temperatur T ausgedrückt werden:


Darin ist die Boltzmann-Konstante. Die Energiedifferenz entspricht dabei der Energie eines Energiequants (), das ein Teilchen vom günstigeren in den ungünstigeren Zustand befördert (Grundgleichung der Spektroskopie). Bei einer Resonanzfrequenz von 600 MHz und einer Temperatur von 0 °C (273 K) ergibt sich ein Wert von ungefähr e−0,0001, also sehr nahe bei eins. Daher sind schon im thermischen Gleichgewicht fast gleich viele Kerne im angeregten Zustand wie im Grundzustand. Zum Vergleich: Sichtbares Licht besitzt um einen Faktor von etwa 1 Million höhere Frequenzen. Folglich haben Übergänge, die durch sichtbares Licht angeregt werden, Besetzungsunterschiede von etwa e−100, liegen also vollständig im Grundzustand vor, was die Spektroskopie im sichtbaren Bereich wesentlich empfindlicher macht.


Die Empfindlichkeit ist somit im Wesentlichen von vier Faktoren abhängig:
- Temperatur (Abhängigkeit ),
- Unterschied der Energiezustände (proportional zu Magnetfeldstärke und isotopspezifischem gyromagnetischen Verhältnis ),
- Häufigkeit des Isotops,
- Spinquantenzahl des betrachteten Isotops.

| NMR-Eigenschaften von Wasserstoff |
|---|
Die Faktoren und die Spinquantenzahl lassen sich durch die relative Empfindlichkeit ausdrücken. Dabei wird 1H als Referenz verwendet. Somit ergibt sich für ein Isotop mit Spinquantenzahl und dem gyromagnetischen Verhältnis bei gleicher Temperatur, gleichem magnetischen Feld und gleicher Isotopenhäufigkeit die relative Empfindlichkeit :



Multipliziert man diesen Wert mit der natürlichen Häufigkeit des Isotops, erhält man die absolute Empfindlichkeit .[8] Seltener wird die relative Empfindlichkeit zu 13C als Referenz angegeben.

Um die Empfindlichkeit zu steigern, werden verschiedene Maßnahmen ergriffen:
- Messung möglichst empfindlicher Nuklide (besonders 1H),
- Anreicherung magnetischer Nuklide (wie 13C oder 15N), deren natürliche Häufigkeit gering ist (z. B. in Proteinen),
- Signal-Akkumulation durch mehrfache Messung einer Probe und Addition aller Spektren,
- Erhöhung der Magnetfeldstärke durch Einsatz stärkerer (supraleitender) Magnete,
- Senkung der Temperatur der Probe,
- Verringerung des elektronischen Rauschens durch Kühlung der Empfänger (Kryoelektronik),
- Verwendung von Hyperpolarisationsmethoden, um die Besetzungsunterschiede künstlich zu vergrößern,
- Ausnutzen des Kern-Overhauser-Effekts (NOE),
- Übertragung der Magnetisierung empfindlicher Kerne (1H) auf unempfindlichere (13C) (Kreuzpolarisation CP).
Die Senkung der Temperatur und die Erhöhung der Magnetfeldstärke ändern das thermische Besetzungsgleichgewicht der -Zustände, so dass mehr Kernspin-Übergänge angeregt werden können. Mit Hilfe der Hyperpolarisation kann ein Besetzungs-Ungleichgewicht erzeugt werden, das stark vom thermischen Gleichgewicht abweicht und in dem der energetisch günstigste -Zustand fast vollständig besetzt ist.
Zur Optimierung der Signaldetektion wird rauscharme Elektronik verwendet. Der Einsatz von elektrischen Schwingkreisen begrenzt die Detektion auf ein schmales Frequenzband im Bereich der erwarteten Larmor-Frequenz, d. h. die Detektion von Störsignalen und von Rauschen aus anderen Frequenzbereichen wird unterdrückt.
Die Signal-Akkumulation dient dazu, das Signal-Rausch-Verhältnis zu verbessern. Eine NMR-Messung wird -mal auf identische Weise durchgeführt und die gemessenen Signale der einzelnen Messungen werden addiert. Durch diese Akkumulation nimmt die NMR-Signalstärke um den Faktor zu, während statistisches Rauschen nur um den Faktor zunimmt. Da zwischen NMR-Experimenten die vollständige -Relaxation der Spins erfolgen sollte und die -Relaxation organischer Substanzen einige zehn Sekunden dauern kann, kann die Signal-Akkumulation zu einer erheblichen Verlängerung der Messdauer führen.


Für typische Messungen sind je nach Experiment und Messzeit ca. 10 nmol bis 1 µmol Substanz notwendig (typische Probenmenge: 1 mL einer Lösung mit einer Konzentration von 10 µmol/L bis 1 mmol/L).
Auflösungsvermögen
Die erreichbare Auflösung eines Pulsspektrometers ist invers proportional zur Länge des FID-Signals. Neben der transversalen Relaxation () der Probe wird sie von der Inhomogenität des -Feldes in der Probe bestimmt. Das Ausgleichen von Magnetfeld-Inhomogenitäten erfolgt über das sogenannte Shimming. Dazu werden im Spektrometer mit Hilfe elektrischer Ströme schwache Magnetfelder zusätzlich zum Hauptmagnetfeld erzeugt, mit denen lokale Inhomogenitäten zum Teil ausgeglichen werden können. Die erhöhte Homogenität des resultierenden Gesamtmagnetfeldes reduziert die -Relaxation, wodurch das NMR-Signal langlebiger wird.
Um höchste Auflösung im Spektrum zu erhalten, d. h. schmale (scharfe) NMR-Linien und Unterscheidbarkeit eng benachbarter Linien, muss eine weitere Technik angewendet werden, nämlich die schnelle Rotation (in engl. spinning) der Messzelle um ihre Längsachse, mit Hilfe einer kleinen Luftturbine. Durch diese makroskopische Bewegung der Probe erfahren die Kerne in der Probe einen zeitlichen Mittelwert des äußeren Feldes über das Probevolumen. Die nach dem Shimmen verbliebenen Magnetfeld-Inhomogenitäten werden also ausgemittelt. So erreicht man schließlich eine Frequenzauflösung in der NMR von ca. 0,1 Hz, was die Unterscheidbarkeit von extrem kleinen Energiedifferenzen bedeutet, die dann eine Fülle von Details aus hochaufgelösten NMR-Spektren liefern können.
Anwendungsgebiete
NMR-Spektren können am einfachsten für Moleküle aufgenommen werden, die sich in Lösung befinden und nicht mit paramagnetischen Substanzen in Wechselwirkung stehen. NMR-Spektroskopie an paramagnetischen Substanzen und an Festkörpern ist ebenfalls möglich, die Interpretation der Spektren und die Aufbereitung der Proben für die Messung sind aber in beiden Fällen deutlich komplexer. Bezüglich der NMR an Festkörpern vgl. auch Magic-Angle-Spinning.
Die hochauflösende Kernresonanzspektroskopie in Lösung wird heute in großem Maßstab für folgende Aufgaben verwendet:
- zum zerstörungsfreien Nachweis von Inhaltsstoffen einer Probe,
- zur Bestimmung von Molekülstrukturen (von kleinen Molekülen bis hin zu Proteinen und Nukleinsäurefragmenten),
- zur Untersuchung von Wechselwirkungen zwischen Molekülen.
Neben spektroskopischen Untersuchungen vermittelt die Bestimmung von Kernspin-Relaxationszeiten Informationen über die Struktur und Dynamik von Materialien. In Flüssigkeiten z. B., deren Mikrostruktur und -Dynamik mit herkömmlichen Methoden nur schwer erforscht werden kann, können Abstände zwischen molekularen Nachbarn und molekulare Umorientierungszeiten, die typischerweise im Pico- bis Nanosekunden-Bereich liegen, mittels Relaxationszeitmessungen bestimmt werden.
Unterschiedliche Kernspin-Relaxationszeiten in verschiedenen biologischen Geweben bilden die Basis für die in der Medizin als bildgebendes diagnostisches Verfahren genutzte Magnetresonanztomographie (Kernspintomographie). Eine Anwendung der kernmagnetischen Resonanz, welche für die Neurowissenschaften, wie Neurologie und Neuropsychologie, eine außerordentliche Bedeutung erlangt hat, ist die funktionelle Magnetresonanztomographie. Die NMR-spektroskopische Messung von Metabolitenkonzentrationen in vivo wird als Magnetresonanzspektroskopie bezeichnet. Magnetresonanztomographie-Methoden finden allgemein, außer in der medizinischen Diagnostik, auch zunehmend Anwendungen in den Ingenieur- und Geowissenschaften.
Ein weiteres wichtiges Anwendungsgebiet ist die Untersuchung der translatorischen Moleküldynamik, also von Diffusion und Fließbewegungen von Molekülen oder Molekülaggregaten in Flüssigkeiten und Festkörpern mittels Feldgradienten-NMR.[9] Mit der sogenannten diffusion-ordered-spectroscopy (DOSY) kann in Mischungen die translatorische Beweglichkeit der, NMR-spektroskopisch identifizierten, Einzelkomponenten gemessen werden. Zusätzlich kann mit Hilfe des DOSY-NMR Experiments das Molgewicht von Molekülen in gelöster Form bestimmt werden.[10][11]
Methoden zur Strukturaufklärung in der organischen Chemie
Chemische Verschiebung und Integration von Signalen

Die Resonanzfrequenzen werden nicht als Absolutwerte, sondern als chemische Verschiebung gegenüber einer Referenzsubstanz, dem so genannten Standard, angegeben. Die chemische Verschiebung ist definiert als
- ,

wodurch sie unabhängig von der Feldstärke des gerade verwendeten Magneten wird. Da die Werte der chemischen Verschiebung sehr klein sind, werden sie in ppm angegeben. Als Standard für 1H- und 13C-Spektren organischer Lösungen wird die Resonanzfrequenz der jeweiligen Kerne in Tetramethylsilan (TMS) verwendet. Während früher jeder Probe einige Milligramm TMS zugesetzt wurden, bezieht man sich heutzutage in der Regel auf die bekannte chemische Verschiebung der Restprotonen des deuterierten Lösungsmittels.
Die chemische Verschiebung von Wasserstoffkernen in organischen Molekülen wird durch die Art der funktionellen Gruppen beeinflusst. Je nach der Struktur des Moleküls weichen die chemischen Verschiebungen gleicher funktioneller Gruppen leicht voneinander ab, so dass das NMR-Spektrum charakteristisch für eine Substanz ist. Außerdem werden sie durch benachbarte Moleküle in der Probe beeinflusst, so dass in unterschiedlichen Lösungsmitteln oder in der Reinsubstanz unterschiedliche relative und absolute Resonanzfrequenzen der Protonen einer Probe auftreten. Bei starken Wechselwirkungen zwischen Substanz und Lösungsmittel treten dabei Unterschiede von mehreren ppm auf. Mit Hilfe der Shoolery-Regel kann die chemische Verschiebung abgeschätzt werden.
Die relative Anzahl der einem bestimmten Signal zugrundeliegenden Wasserstoffatome ist bei einfacher 1H-Spektroskopie proportional zum Flächeninhalt (dem Integral) des betreffenden Signals.
Durch Auswertung dieses Integrals kann also beispielsweise bestimmt werden, wie viele Wasserstoffatome eines Moleküls sich an Methylgruppen, an Aromaten, an Carboxygruppen, an Doppelbindungen usw. befinden. Diese Kenntnis ist für die organische Chemie bei der Bestimmung von Strukturen äußerst wichtig. Eine Übersicht zur Zuordnung bestimmter funktioneller Gruppen (Atomgruppen, Stoffgruppen) zu Werten der chemischen Verschiebung bietet die folgende Tabelle.
| Wasserstoffatome | typische chemische Verschiebungen [ppm] |
|---|---|
| H3C– | 0,9 |
| H3C-Vinyl-R3 | 1,6 |
| H3C–Ar | 2,3 |
| H3C–CO–R | 2,2 |
| H3C–OR | 3,3 |
| R2C–CH2–CR2 | 1,4 |
| –C–CH2–Cl | 3,6 |
| Ar-H | 6,8 bis 7,5 (bis 8,5 bei Heteroaromaten) |
| R-CHO | 9 bis 10 |
| R–COOH | 9 bis 13 |
| ROH | 0,5 bis 4,5 |

Skalare Kopplung
Spinzustände werden durch weitere Kerne, die sich in ihrer Nachbarschaft befinden und von Null verschiedenen Spins haben, in energetisch unterschiedliche Niveaus aufgespalten. Die Zahl dieser Niveaus hängt ab von der Anzahl möglicher unterschiedlicher Orientierungen der einzelnen Spins. Diese skalare Kopplung wird durch die Spins der Elektronenpaare vermittelt, welche die Bindung zwischen den Teilchen bilden; ihre Wirkung ist gewöhnlich über bis zu vier Bindungen feststellbar. Der Abstand der Linien ist unabhängig vom angelegten Feld und wird deswegen als absoluter Frequenzunterschied (in Hertz) angegeben. Bei Kernen, die chemisch und spin-symmetrisch gleich (magnetisch äquivalent) sind, ist die Kopplung nicht sichtbar.
Aus dem beobachteten Kopplungsmuster kann der Spektroskopiker die Nachbarschaftsverhältnisse der einzelnen Kerne und damit in vielen Fällen die vollständige Struktur einer Verbindung erschließen.
Mitunter kann es hilfreich sein, für die Strukturaufklärung eine bestimmte oder alle Kopplungen zu unterdrücken. Dazu wird ein Radiosignal mit der Frequenz eines Kernes oder einer ganzen Gruppe von Kernen (Breitbandentkopplung) eingestrahlt; das übrige Spektrum verhält sich dann so, als wäre der entsprechende Kern nicht vorhanden. 13C-Spektren werden standardmäßig 1H-entkoppelt, da sie durch die Überlappung der Kopplungsmuster der einzelnen Kerne sonst oft uninterpretierbar wären. Außerdem wird die geringe Empfindlichkeit der Methode durch die fehlende Aufspaltung verbessert.
In festen oder hochviskosen Proben wird der richtungsabhängige Teil der skalaren Kopplung sowie die dipolare Kopplung nicht mehr zu null ausgemittelt. Solche Proben zeigen große, feldunabhängige Aufspaltungen bzw. Linienverbreiterungen von mehreren Kilohertz für 1H-Spektren.
Organischer NMR-Spektrensatz
Der organische NMR-Spektrensatz umfasst sechs – im Fall eines vorhandenen und leicht messbaren Heteroatoms (z. B. 31P oder 19F) – sieben NMR-Experimente, die (insbesondere in Verbindung mit Daten aus Massenspektrometrie, IR-Spektroskopie und UV/VIS-Spektroskopie) besonders hilfreich zur Aufklärung der Struktur einer organischen Verbindung sind und deshalb in der Regel als erste durchgeführt werden, um eine Struktur aufzuklären.[12] Dies sind, in der Reihenfolge ihrer Bedeutung absteigend:[13]
- 1H-NMR
- 1H-COSY (correlation spectroscopy)
- 13C-DEPT, 13C-INEPT oder 13C-APT (distortionless enhancement by polarization transfer, insensitive nuclear enhancement by polarisation transfer bzw. attached proton test)
- beispielsweise 31P- oder 19F-NMR (heteronukleare Spektren, falls ein Heteroatom vorhanden ist)
- 1H-13C-HSQC oder 1H-13C-HMQC, (heteronuclear single quantum coherence) bzw. (heteronuclear multiple quantum coherence)
- 1H-13C-HMBC (heteronuclear multiple bond correlation)
- 1H-NOESY (nuclear Overhauser effect spectroscopy) als 1D oder 2D-Variante
Weitere NMR-Experimente, die zur weiteren Strukturaufklärung von organischen Verbindungen verwendet werden, sind:
- 1H-TOCSY (total correlaction spectroscopy)
- 13C-INADEQUAT (incredible natural abundance double quantum transfer spectroscopy)
- 13C-ADEQUAT (adequate sensitivity double quantum transfer spectroscopy)
- 1D/2D-DOSY (diffusion ordered spectroscopy)
- 1D/2D-ROESY (rotating frame nuclear Overhauser effect spectroscopy)
- 1D/2D-HOESY (heteronuclear Overhauser effect spectroscopy)
- EXSY (exchange spectroscopy)
Funktionsprinzip
Die Kernsuszeptibilitäten sind wesentlich geringer als die diamagnetischen und paramagnetischen Suszeptibilitäten (Faktor:104). Bei Dia- und Paramagnetismus sind die Elektronen des Atoms für die Suszeptibilität verantwortlich. Bei Kernen kann die Suszeptibilität mit der Langevin-Debye-Formel bestimmt werden.
Früher wurden NMR-Resonanzen mit einer Brückenschaltung in Schwingkreisen bestimmt. Bloch und Mitarbeiter nutzten zwei identische Schwingkreise, d. h. zwei Spulen und zwei Kondensatoren, um einen Abgleich mit einer Brückenschaltung vorzunehmen; eine Spule als Sender eine als Empfänger. Es ist möglich, eine Brückenschaltung mit nur einer Spule herzustellen. Dieses Verfahren wurde von Purcell genutzt.
Vor der Probenvermessung wird die Brücke mit der zu messenden Frequenz abgeglichen. Mit Gleichungen aus der Physik kann man für einen Schwingkreis und eine Brückenschaltung die Phasenverschiebung zwischen Strom und Spannung, den Scheinwiderstand und die Stromlosigkeitsbedingungen einer Brücke berechnen.
In die Spule kommt nun ein Substanzröhrchen hinein. Ein Magnetfeld (mit einem Permanentmagneten oder Elektromagneten) wird dann horizontal zur Spulenachse erzeugt. Bei einer ganz bestimmten Frequenz und einer bestimmten Magnetfeldstärke und nur bei Anwesenheit einer Substanzprobe (mit entsprechenden Atomkernen) wird der Schwingkreis verstimmt. Im Oszilloskop oder mit einem Schreiber ist diese Verstimmung sichtbar.
Sehr bedeutsam war die Bestimmung der räumlichen Magnetisierung durch das angelegte Magnetfeld. Bloch führte für alle Raumrichtungen Berechnungen durch und konnte die schwingungsabhängigen Suszeptibilitäten für die Raumrichtungen ableiten (Bloch-Gleichungen). Ungeklärt blieb jedoch noch die Frage der Relaxationszeit, das heißt, der Zeitdauer bis der angeregte Kernspin auf das Grundniveau zurückfällt. Mittels paramagnetischer Salze konnte dann die Relaxationszeit von reinen Wasserstoffprotonen auf etwa drei Sekunden berechnet werden.
Wasserstoffkerne konnten bei sehr geringen Frequenzen (wenige Kilohertz) und einem sehr schwachen Magnetfeld durch Schwingkreisverstimmung nachgewiesen werden. Interessant wird die Methode für die Strukturaufklärung von komplexen Molekülen jedoch erst bei hohen Frequenzen (ab 60 MHz) und stärkeren Magnetfeldern (1,4 Tesla), da sich dann die chemischen Verschiebungen von unterschiedlichen Wasserstoffatomen komplizierter Verbindungen deutlicher unterscheiden. Will man jedoch nicht nur ein einziges Signal auf dem Oszilloskop sehen, sondern mehrere unterschiedliche Wasserstoffatomkerne (oder andere Kerne) so muss ein ganzes Frequenzband eingestrahlt werden.
Früher – bis in die 1970er Jahre – nutzten die NMR-Spektrometer das Continuous-Wave-Verfahren (CW), um das Spektrum einer komplexen Verbindung abzutasten.
Heute ist die Puls-Fourier-Transformation (PFT) üblich. Hierbei wird ein Hochfrequenzimpuls eingestrahlt. Dieser Impuls enthält ein ganzes Band an Schwingungen.
Die bereits angesprochene Abhängigkeit der Energieniveaus der Kernspins von der Molekülstruktur rührt in erster Linie von der Wechselwirkung der Elektronenstruktur der Moleküle mit dem äußeren Magnetfeld her: Hierdurch entsteht in der Elektronenhülle ein Induktionsstrom, welcher wiederum ein Magnetfeld erzeugt, das dem äußeren entgegengerichtet ist. Dadurch wird das Magnetfeld am Atomkern geschwächt, die Frequenz der für den Übergang notwendigen Strahlung ist also kleiner als im Falle eines nackten Atomkerns. Die Differenz heißt chemische Verschiebung und wird üblicherweise im Verhältnis zur für den nackten Atomkern nötigen Frequenz angegeben. Chemische Verschiebungen liegen üblicherweise im Bereich von 0–5000 ppm.
Das magnetische Feld wird am Atomkern durch die Ausrichtung weiterer magnetischer Momente in der unmittelbaren Umgebung beeinflusst. Befindet sich beispielsweise ein Kern mit zwei Ausrichtungsmöglichkeiten in der Nähe, so kann dieser das Feld verstärken oder abschwächen. Dies führt zu einer Aufspaltung des Signals, man spricht von einer Kopplung. Weil die chemische Verschiebung im Wesentlichen von der Elektronendichte am Atomkern abhängt, kann man für Atomkerne in chemisch ähnlichen Umgebungen ähnliche Verschiebungen erwarten. Aus der Kopplung erhält man zusätzlich Informationen über Nachbarschaftsbeziehungen zwischen verschiedenen Kernen in einem Molekül. Beides zusammengenommen liefert wesentliche Hinweise über die Struktur des gesamten Moleküls.
Atomkerne mit einer ungeraden Protonen- und/oder Neutronen-Zahl besitzen einen Kernspin I. Dieser kann ganz- und halbzahlige Werte (0, 1⁄2, 1, 3⁄2, 2, 5⁄2, …) annehmen: bei den sogenannten uu-Kernen ist I = n (also nur ganzzahlig: 0, 1, 2, 3, …) während bei gu- und ug-Kernen I = (2n+1)/2 ist (also halbzahlig: 1⁄2, 3⁄2, 5⁄2, …), bei Isotopen mit gerader Protonen- und Neutronenzahl (sogenannten gg-Kernen) ist I = 0. Von Null verschiedene Kernspins gehen mit einem magnetischen Dipolmoment einher. Die Größe dieses Dipolmoments wird durch das gyromagnetische Verhältnis des betreffenden Isotops beschrieben. In einem äußeren, statischen Magnetfeld richten sich magnetische Kernmomente entsprechend den Regeln der Quantenmechanik aus. Ein Atomkern mit I = ½ hat die Form einer Kugel, Kerne mit I > ½ haben eine ellipsoidische Form und haben daher zusätzlich ein elektrisches Quadrupolmoment „eQ“, welches mit elektrischen Feldgradienten wechselwirken kann (siehe auch Kernquadrupolresonanz-Spektroskopie). Diese zusätzliche starke, elektrische Wechselwirkungsmöglichkeit führt zu breiten NMR-Resonanzlinien, die komplizierter zu interpretieren sind als die schmalen, durch gut auflösbare Kopplungen strukturierten Resonanzlinien der Spin-½-Kerne.
Die am meisten für die chemische Strukturaufklärung genutzten Isotope sind daher Kerne mit Spin ½. Hierzu gehören unter anderem die Nuklide 1H, 13C, 15N, 19F, 29Si und 31P. Spin-½-Kerne können nur zwei diskrete Zustände annehmen, nämlich entweder parallel oder antiparallel zum äußeren Magnetfeld. Zwischenstellungen sind quantenmechanisch verboten. Die zwei Anordnungsmöglichkeiten entsprechen zwei unterschiedlichen Energiezuständen.
Die Energiedifferenz zwischen diesen beiden Zuständen ist proportional zur Stärke des Magnetfelds am Kernort. Der Proportionalitätsfaktor ist dabei das gyromagnetische Verhältnis des betreffenden Isotops. Übergänge zwischen den beiden Orientierungen der Kernmomente können durch die Einstrahlung resonanter magnetischer Wechselfelder ausgelöst werden. Die Resonanzfrequenz ist der Energieaufspaltung zwischen den beiden Kernspins proportional und wird als Larmorfrequenz bezeichnet.

Veranschaulichen lässt sich dies durch das nebenstehende Diagramm. Hierbei denkt man sich ein Koordinatensystem mit dem äußeren Magnetfeld entlang der z-Achse. Ein Atomkern mit einem Spin von ½ richtet sich mit einem Spin-Vektor entweder parallel oder antiparallel zum äußeren Feld aus. Wenn man nun die Vektoren mehrerer Atome in dieses Koordinatensystem aufnimmt, entstehen zwei Kegel, jeweils einer für parallel und antiparallel. Infolge des Energie-Unterschieds zwischen der parallelen und der antiparallelen Orientierung der magnetischen Kernmomente gibt es im thermischen Gleichgewicht einen Besetzungsunterschied zwischen den beiden Orientierungen. Dieser folgt in Hochtemperatur-Näherung der Boltzmann-Verteilung und bewirkt eine Überschussmagnetisierung in positiver Richtung entlang der z-Achse.
Das NMR-Signal kommt dadurch zustande, dass man die zu untersuchende Probe im Magnetfeld einem Radiofrequenz-Puls aussetzt. Dabei werden die Spins der einzelnen Atome durch das Magnetfeld des Pulses beeinflusst, so dass der Gesamtvektor, der sich aus den gezeigten Spin-Kegeln ergibt, gekippt wird. Er liegt nun nicht mehr parallel zur z-Achse, sondern ist um einen Winkel ausgelenkt, der proportional zur Dauer und Intensität des Radiofrequenzpulses ist. Typisch sind Pulslängen von etwa 1–10 µs. Eine maximale Quermagnetisierung senkrecht zur z-Achse wird bei einem Auslenkungswinkel von 90° erreicht.
Diese Quermagnetisierung verhält sich wie ein magnetischer Kreisel und präzediert in der Ebene senkrecht zum statischen Magnetfeld. Diese Präzessionsbewegung macht sich als sehr schwaches magnetisches Wechselfeld bemerkbar, das mittels geeigneter Verstärker gemessen wird. Nach Beenden der resonanten Einstrahlung treten zwei Prozesse, sogenannte Relaxationsprozesse, ein, durch die die Quermagnetisierung wieder abnimmt. Das NMR-Signal wird also nach Beenden des Radiofrequenzpulses als Freier Induktionszerfall (FID; von englisch: free induction decay) gemessen. Die Zeitkonstante dieses freien Induktionszerfalls hängt von der transversalen Relaxationszeit sowie von der Homogenität des Magnetfelds ab. Für leicht bewegliche Flüssigkeiten in homogenen Magnetfeldern kann sie im Bereich von mehreren Sekunden liegen. Der FID wird durch die Frequenzunterschiede infolge von chemischer Verschiebung und Kopplung moduliert. Durch eine Fourier-Transformation kann die Verteilung der verschiedenen Frequenzen aus dem FID berechnet werden. Dies ist dann das NMR-Spektrum. Das NMR-Spektrum liefert in vielen Fällen einen eindeutigen „Fingerabdruck“ des jeweiligen Moleküls. Zusammen mit Informationen aus weiteren Messungen wie z. B. der Massenspektrometrie kann aus den Spektren die chemische Struktur einer unbekannten Substanz bestimmt werden.
Kommerzielle NMR-Spektrometer für die chemische Strukturaufklärung arbeiten üblicherweise bei magnetische Flussdichten zwischen 7 und 21 Tesla. Für 1H entsprechen die Resonanzfrequenzen (Larmorfrequenzen) dann zwischen 300 und 900 MHz. Da 1H der wichtigste NMR-Kern ist, wird die Feldstärke von Spektrometern gewöhnlich in dessen Larmorfrequenz ausgedrückt. Bei 1H beträgt die Aufspaltung der Spektren infolge unterschiedlicher chemischer Verschiebungen ca. 10 ppm. Dies entspricht also einer maximalen Bandbreite von ca. 3 kHz bei einer NMR-Frequenz von 300 MHz. Die Frequenzbandbreite der NMR-Spektren infolge der chemischen Verschiebung wächst proportional zum Magnetfeld an. Die chemische Verschiebung selbst ist als Verhältnis der Differenz der Resonanzfrequenz des Kerns in einer bestimmten chemischen Umgebung und der Resonanzfrequenz in einer Referenzverbindung zur Resonanzfrequenz selbst definiert. Dies erlaubt einen einfachen Vergleich zwischen NMR-Spektren, die bei verschiedenen Feldern gemessen wurden. Für Wasserstoff und Kohlenstoff wird Tetramethylsilan (TMS) als Referenzsubstanz genommen. Der Bereich von chemischen Verschiebungen für Kohlenstoff und viele andere Kerne ist wesentlich breiter als für Wasserstoff und kann mehrere 100 ppm betragen. Bei einigen sehr schweren Kernen wie z. B. 207Pb werden chemische Verschiebungen im Bereich von Prozent beobachtet.
Puls-Fourier-Transform NMR
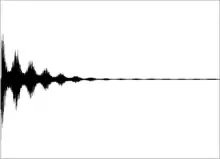
Heutzutage arbeiten alle modernen NMR-Spektrometer mit der Puls-Technik. Bei dieser wird ein einzelner Radiofrequenzimpuls (RF-Puls) oder eine Sequenz von RF-Pulsen auf eine Probe gesandt, die sich in einem starken Magnetfeld befindet. Nach dem Abklingen des Pulses in der Empfangselektronik (Totzeit) wird der Zerfall der Magnetisierung (engl. free induction decay, FID), d. h., ihre Rückkehr in den Gleichgewichtszustand, über die dadurch in der Empfangsspule induzierte Spannung als Funktion der Zeit detektiert. Durch Fourier-Transformation wird dieses Zeitsignal im Computer in das Frequenzspektrum (Signalintensität als Funktion der Frequenz) transformiert.
Diese Messtechnik hat das früher verwendete CW-Verfahren (engl. continuous wave) (s. o.) fast vollständig verdrängt.
Experimentelle Größen
- Die chemische Verschiebung einer Resonanz ist vom lokalen Magnetfeld am Kernort abhängig, das wiederum von der chemischen Umgebung des betrachteten Kerns abhängt.
- Die Intensität einer Resonanz ist zumeist proportional zur Anzahl der sie hervorrufenden Kerne, solange die Besetzungsunterschiede nicht durch starke Kopplung oder Suszeptibilitätsunterschiede verändert werden.
- Bei den Relaxationszeiten angeregter Zustände unterscheidet man zwischen longitudinaler Relaxationszeit (Spin-Gitter-Relaxation) und transversaler Relaxationszeit (Spin-Spin Relaxation). Longitudinale Relaxationszeiten bestimmen die Einstellung der Gleichgewichtsmagnetisierung. Die transversalen Relaxationszeiten bestimmen die Linienbreite der Resonanzlinien. Relaxationseffekte geben Aufschluss über vorhandene Wechselwirkungen und molekulare Bewegungen.
- Räumlich benachbarte Kerne wechselwirken miteinander über magnetische Dipol-Dipol-Wechselwirkung (dipolare Kopplung). Diese Wechselwirkung verschwindet in isotropen Lösungen im zeitlichen Mittel.
- Indirekt können Kerne über chemische Bindungen miteinander wechselwirken. Diese skalare Kopplung ist für die Aufspaltung der Signale in Multipletts verantwortlich und stellt eine wesentliche Grundlage für die molekulare Strukturbestimmung mit NMR dar. Der Abstand zweier benachbarter Linien eines Multipletts wird als Kopplungskonstante, die in Hertz gemessen wird, bezeichnet.
Eindimensionale NMR-Spektroskopie
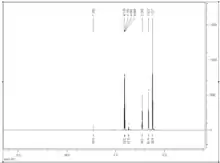
Die eindimensionale NMR-Spektroskopie ist die am häufigsten angewandte Strukturaufklärungsmethode der Chemie. Bei ihr wird die chemische Verschiebung eines Atoms von einer Referenzsubstanz gemessen. 1H und 13C sind die Kerne, die am häufigsten in der organischen Chemie gemessen werden, aber auch 15N, 31P, 19F und viele andere NMR-aktive Isotope können spektroskopiert werden.
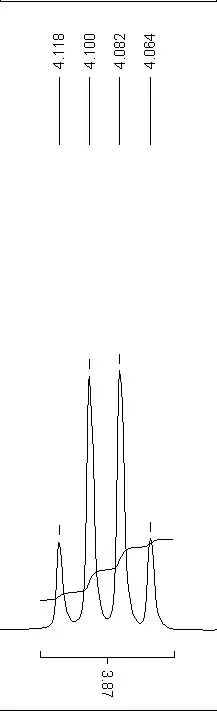
Das Aussehen der Spektren hängt entscheidend von der Aufnahmeart ab. 1H-Spektren werden in der Regel nicht Breitband-entkoppelt aufgenommen. Damit haben alle Wasserstoffatome die Möglichkeit, ihren Spin mit anderen Kernen zu koppeln, die sogenannte Spin-Spin-Kopplung. Damit entsteht bei der charakteristischen chemischen Verschiebung eines Atoms eine für seine Umgebung charakteristische Aufspaltung des Signals, aus der Informationen über die Molekülstruktur abgeleitet werden können.
13C, 15N, 31P, 19F und andere Kerne werden häufig 1H-Breitband-entkoppelt aufgenommen, so dass die Aufspaltung der Signale aufgrund der Kopplungen zu 1H-Kernen ausbleibt.
Skalare Kopplung
Der Kern eines Atoms kann mit einem benachbarten Atomkern in Wechselwirkung treten. Das kann entweder direkt (durch den Raum) oder indirekt (über die Bindungselektronen zwischen den Kernen) geschehen. Bei einer flüssigen Probe mittelt sich die direkte (dipolare) Wechselwirkung durch die schnelle Bewegung der Kerne im Raum aus. Erhalten bleibt die skalare Kopplung, die dadurch vermittelt wird, dass die (stets gepaart (↓↑)) auftretenden Spins der Bindungselektronen unterschiedlich mit den Kernspins auf beiden Seiten der Bindung wechselwirken.
Hat ein Kern den Zustand α (↑), so wird das (↑)-Elektron der Bindung von ihm abgestoßen, hält sich also eher am anderen Kern auf. Weitere von dort ausgehende Bindungen werden gleichfalls (in geringerem Ausmaß) spinpolarisiert. Wird so ein weiterer Kern mit erreicht, ergibt sich ein Energieunterschied zwischen dessen Zustand α und β durch wiederum seine Wechselwirkung mit den Elektronen der Bindung. Solche Kopplungen sind gewöhnlich über max. drei bis vier Bindungen nachweisbar, in konjugierten π-Systemen zum Beispiel aber auch weiter.

Das NMR-Signal des ersten Kerns wird dadurch zu einem Dublett (für , sonst Linien) aufgespalten, und das des zweiten Kerns gleichfalls, und zwar um den gleichen Betrag, da der Energieunterschied (sog. Kopplungskonstante) zwischen αα(=ββ) und αβ(=βα) derselbe sein muss. Durch die gleich starke Aufspaltung ist dann die Nachbarschaft der beiden Atome im Molekül nachgewiesen. Koppelt ein Kern noch zu weiteren anderen, so wird jede seiner Linien entsprechend nochmals aufgespalten.

Koppelt ein Kern zu zwei (oder allgemein ) gleichartigen Nachbarkernen (die aus der Sicht des Ursprungskerns die gleiche chemische Umgebung und Spin-Symmetrie besitzen), so erhält man ein Triplett, da die mittleren Linien des „Dubletts vom Dublett“ (oder Quartett, Quintett usw., also Linien, bzw. Linien für Kerne mit ) zusammenfallen. Die relativen Intensitäten der Linien ergeben sich (für – Kerne) aus der -ten Zeile des Pascalschens Dreiecks, also 1:2:1 oder 1:3:3:1. Sind die koppelnden Kerne nicht gleichartig, d. h. sind ihre Kopplungskonstanten unterschiedlich, so fallen die mittleren Linien nicht zusammen, man erhält dann z. B. ein Dublett vom Triplett o. ä.



Beispielspektren
Als ein einfaches Beispiel dient Propan (H3C–CH2–CH3): Die CH2-Gruppe beim Propan hat zwei benachbarte Methylgruppen (–CH3). Dies entspricht sechs benachbarten, äquivalenten H-Atomen. Das Signal wird also in ein Septett aufgespalten. Die Methyl-Protonen werden von den beiden Methylen-Protonen zum Triplett aufgespalten, die 4J-Kopplung zu den drei anderen Methyl-Protonen ist unsichtbar, da diese magnetisch äquivalent sind, so wie auch keine 2J-Kopplung innerhalb der Methylgruppen beobachtet werden kann.
Sind in einem Molekül mehrere unterschiedliche z. B. Methyl-Gruppen vorhanden, so überlagern sich deren Multipletts häufig, wodurch sie schnell unauswertbar werden. Um solche Fälle besser auflösen zu können, wird hierfür vielfach auf mehrdimensionale NMR-Techniken wie COSY zurückgegriffen. Da die Aufspaltung nicht feldabhängig ist, der Abstand zwischen den Signalen chemisch unterschiedlicher Protonen aber schon, können Überlagerungen durch Anwendung eines höheren Feldes aufgelöst werden.
Erklärungen zum Spektrum von Ethanol:
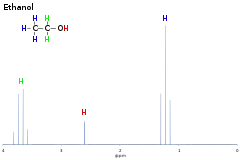

Die OH-Gruppe zeigt nur ein Singulett, wenn das Ethanol der Probe mehr als 3 % Wasser enthält. Das alkoholische Wasserstoffatom ist leicht acid und wird deswegen ständig durch Wasserstoffatome anderer Hydroxygruppen oder aus dem Lösungsmittel ausgetauscht. Dies führt dazu, dass keine permanenten Spin-Spin-Kopplungseffekte zwischen OH-Gruppe und benachbarter Methylengruppe auftreten. Bei sehr tiefen Temperaturen oder in absolut wasserfreiem Alkohol ist dieser Austausch genügend verlangsamt, um dann die erwartete vicinale Kopplung () sichtbar zu machen.[14]

Zweidimensionale NMR-Spektroskopie
Bei der zweidimensionaler Kernspinresonanzspektroskopie (2D-NMR) werden die Intensitäten in Abhängigkeit von zwei Frequenzachsen aufgezeichnet. Sie generiert also dreidimensionale Diagramme. Zweidimensionale NMR-Spektren liefern mehr Informationen über ein Molekül als eindimensionale NMR-Spektren und sind deshalb besonders nützlich bei der Bestimmung der Struktur eines Moleküls, insbesondere für Moleküle deren Struktur zu kompliziert ist, um sie mit eindimensionalen NMR zu untersuchen.
Niederfeld-NMR
Es gibt relativ preiswerte Niedrigfeld-NMR-Geräte (10 … 60 MHz), die, mit einem Permanentmagneten ausgestattet, zwar keine aufgelösten Spektren liefern, dafür aber in den Betriebskosten unvergleichlich günstiger (keine Helium-Kühlung) sind. Auch können solche Systeme portabel ausgelegt werden. Durch Analyse der 1H-Relaxationszeiten können Mischungsanteile von Mehrstoffsystemen (Suspensionen, teilkristalline Substanzen[15]) und, nach Kalibrierung, auch Absolutmengen von Stoffen quantifiziert werden, was besonders in der Industrie interessant ist. Messungen erfolgen dabei anstatt in einem (teuren) deuterierten Lösungsmittel üblicherweise in Substanz. Andere Kerne als Wasserstoff werden aufgrund der geringen Empfindlichkeit nur selten untersucht.
Deuterium-Kernspinresonanzspektroskopie
Deuterium (D, 2H) stellt insofern eine Besonderheit dar, weil der Spin beträgt. Das hat zur Folge, dass die Linienbreite der NMR-Signale gegenüber 1H-Kernen größer ist. Die Resonanzfrequenzen liegen deutlich unter denen von 1H-Kernen (61,4 gegenüber 400 MHz bei 9,39 Tesla). Die Deuterium-NMR-Spektroskopie ist ungefähr um den Faktor 100 unempfindlicher als die 1H-NMR-Spektroskopie. Außerdem ist der Deuteriumanteil gegenüber Wasserstoff in organischen Verbindungen sehr gering (ca. 0,0159 %). Mit modernen NMR-Spektrometern stellt die Untersuchung jedoch heute kein Problem dar. Die Aufnahmen bzw. Auswertungen erfolgen mit der Fourier-Transform-Methode. Die Interpretation der Spektren ist nicht schwierig, weil die chemischen Verschiebungen praktisch identisch mit denen von 1H sind.
Mit der 2H-NMR-Spektroskopie lassen sich für die sog. SNIF-NMR die Deuteriumverteilung in den einzelnen Positionen in einer organischen Verbindung und das D/H-Verhältnis positionsspezifisch bestimmen. Die Deuteriumverteilung lässt sich aus den Spektren direkt ablesen, das D/H-Verhältnis kann man nur bestimmen, wenn man einen Standard mit einem bekannten D/H-Verhältnis benutzt. Diese Methode ist für die Analytik bedeutungsvoll, weil damit eine Aussage über die Herkunft einer organischen Verbindung getroffen werden kann. Dies ist einerseits darin begründet, dass der Deuteriumanteil auf der Erde unterschiedlich ist und die Ausgangsstoffe, für Naturstoffe im Wesentlichen auch Wasser, damit einen geringfügigen Unterschied im Deuteriumgehalt aufweisen. Andererseits ist bei den Synthesewegen der kinetische Isotopeneffekt von Bedeutung. So weist z. B. Ethanol in Weinen aus unterschiedlichen Regionen eine unterschiedliche Deuteriumverteilung im Molekül bzw. ein unterschiedliches D/H-Verhältnis auf. Das Gleiche gilt für alle Naturstoffe und somit kann man inzwischen für viele dieser Stoffe eine Herkunft beziehungsweise die Syntheseart, ob natürlich oder synthetisch, zuordnen.[16]
Bei über Gärungsprozesse aus Zucker hergestelltem Ethanol lässt sich über das D/H-Verhältnis die pflanzliche Herkunft feststellen, also ob der Zucker aus Zuckerrohr, Rüben, Getreide, Mais, Kartoffeln, Trauben oder Äpfeln stammt. Somit lassen sich unerlaubte Zuckerzusätze ermitteln. Neben Ethanol können zudem aus 13C-Spektren weitere Inhaltsstoffe im Wein, wie Glycerin, Methanol, Organische Säuren und Konservierungsmittel qualitativ und quantitativ bestimmt werden.[17]
Weiterhin ist Deuterium-NMR eine verbreitete Methode zur Untersuchung einer Orientierungsanisotropie in hochmolekularen organischen Festkörpern (z. B. Fasern).
Metallkern-Kernspinresonanzspektroskopie
Neben den Untersuchungen von organischen Verbindungen mit der 1H-, 13C- und 19F-NMR-Spektroskopie ist die Metallkern-NMR-Spektroskopie von Bedeutung. Hierbei können Metall-Metall- bzw. Metall-Ligand-Bindungen in Komplexverbindungen und metallorganischen Verbindungen direkt untersucht werden. Auch lassen sich Proteine mit eingelagerten Metallionen untersuchen. Kurzlebige Zwischenstufen, die nach der Reaktion nicht oder nur schwer nachweisbar sind, lassen sich in den Reaktionslösungen nachweisen. Man benötigt keine speziellen Lösungsmittel, sondern kann die Messungen in der Reaktionslösung durchführen. Beispiele sind die 6/7Li-, 25Mg-, 27Al-NMR-Spektroskopie und Messungen an Schwermetallkernen, wie z. B. 195Pt, 205Tl und 207Pb.
Die Untersuchung elektrisch leitender metallischer Festkörper erfordert andere experimentelle Voraussetzungen als die von Metallkernen in Lösungen. Die Kernspin-Quantenzahl mancher Metallkerne ist größer als 1/2 (Beispiele: 6Li: 1, 7Li: 3/2, 23Na: 3/2, 25Mg: 5/2, 59Co: 7/2). Solche Kerne haben ein elektrisches Quadrupolmoment, sie relaxieren über einen besonderen Relaxationsmechanismus und haben daher oft sehr breite Resonanzlinien, was Auswirkungen auf die Empfindlichkeit der NMR-Messungen hat.[18]
Hersteller
Pionier war die Firma Varian Associates in Palo Alto, Kalifornien. Varian wurde 1948 gegründet und fertigte hochauflösende Flüssigkeitsspektrometer, die auf Continuous-Wave-Sweep-Methoden und Elektromagneten basierten. Bedeutende Produkte waren: R30 (1952), HR40 (1955), HR60 (1958), HR100 (1959). Mit dem Erfolg des A-60-Spektrometer (1961) wurde Varian weltweiter Marktführer.
Das japanische Unternehmen JEOL stellte im Jahre 1956 ein kommerzielles NMR-System vor, das 1H-Signale bei 32 MHz aufnehmen konnte.
Die Firma Trüb, Täuber & Co. AG, Zürich entwickelte in den 1960er-Jahren in enger Zusammenarbeit mit der ETH Zürich NMR-Spektrometer und baute die Geräte KIS-1 und KIS-2 (1963) als Kleinserie. Die Firma wurde 1965 geschlossen.
Die Firma Bruker wurde 1960 in Karlsruhe gegründet und fertigte Hochleistungs-Magnete, später auch NMR-Spektrometer. Bruker übernahm 1965 die Forschungsabteilung der Firma Trüb und Täuber und gründete damit die Firma Spectrospin AG, Zürich. Durch diese Synergie entstand 1967 das hochauflösende HFX-90 Spektrometer mit 90 MHz, das erste volltransistorisierte NMR-Spektrometer. 1972 entstand bei Spectrospin der erste reine FT Spektrometer, das WH 90 System, der Urahn aller modernen Kernresonanzspektrometer. 2010 hatte Bruker einen weltweiten Marktanteil in der Kernresonanzspektroskopie von etwa 80 %.[19]
Siehe auch
Literatur
- Malcom H. Lewitt: Spin Dynamics. Wiley & Sons, Chichester 2001, ISBN 0-471-48922-0.
- Manfred Holz, Bertold Knüttel: Gepulste Kernspinresonanz: Der heutige Stand einer physikalischen Methode mit einer Vielzahl von Anwendungsmöglichkeiten. In: Physik Journal. Band 38, Nr. 12, Dezember 1982, S. 368–374, doi:10.1002/phbl.19820381208.
- Harald Günther: NMR-Spektroskopie. 3. Auflage. Thieme, Stuttgart 1992, ISBN 3-13-487503-9.
- Ullmanns Enzyklopädie der Technischen Chemie. Band 5, 4. Auflage. Weinheim 1980, ISBN 3-527-20005-3, S. 382 ff.
- A. Streitwieser Jr., C. H. Heathcock: Organische Chemie. Verlag Chemie, Basel 1980, ISBN 3-527-25810-8, S. 205–249.
- Dudley H. Williams, Ian Fleming: Spektroskopische Methoden zur Strukturaufklärung, Kernmagnetische Resonanz-Spektren. Thieme, Stuttgart 1975, ISBN 3-13-437203-7, S. 80–161.
- H. Friebolin: Ein- und Zweidimensionale NMR-Spektroskopie. 4. Auflage. Wiley-VCH, Weinheim 2006, ISBN 3-527-31571-3.
- Stefan Berger, Siegmar Braun: 200 and More Basic NMR Experiments. A Practical Course. Wiley-VCH, Weinheim 2004, ISBN 3-527-31067-3.
- James Keeler: Understanding NMR Spectroscopy. 2. Auflage. Wiley & Sons, Chichester 2010, ISBN 978-0-470-74608-0.
- Timothy D. W. Claridge: High-Resolution NMR Techniques in Organic Chemistry. 2. Auflage. Elsevier, Oxford 2009, ISBN 978-0-08-054818-0.
Weblinks
- TU Dortmund: NMR-Spektrometer (PDF; 2,1 MB; nicht mehr öffentlich zugänglich, erfordert Authentifizierung)
- Verzeichnis der ETH Zürich von Datenbanken und Nachschlagewerken mit 1H-Spektren
- Verzeichnis der ETH Zürich von Datenbanken und Nachschlagewerken mit 13C-Spektren
- Leicht verständliche Shockwave-Animation des NMR-Prinzips – University of Florida
- Spektrendatenbank (nicht nur für NMR) (englisch)
- NMR-Spektrendatenbank (englisch)
- Kernspinresonanz – Einführung in die Theorie. Enthält viel Werbung und tote Links.
- Basics of NMR von Joseph P. Hornak, Ph. D. (englisch)
- e-MRI –MRI step-by-step, interactive course on magnetic resonance imaging, von Campus Medica
- 1H-NMR Prediction – Onlineberechnung von 1H NMR Spektren (englisch)
- 1H-NMR ohne Formeln – Einführung für den Schulunterricht
- James Shoolery: A Basic Guide to NMR, 3rd Edition, Stan's Library, Vol.II, 2008, doi:10.3247/sl2nmr08.012 (englisch; PDF; 4,15 MB)
- nmr.ch – Swiss NMR Community (englisch)
- Aufgabensammlung – NMR-Probleme unterschiedlichen Schwierigkeitsgrades mit Musterlösung als PDF-Download
Einzelnachweise
- Continuous-Wave Nuclear Magnetic Resonance (NMR) Spectroscopy. In: adelaide.edu.au. Department of Chemistry, University of Adelaide, archiviert vom Original am 25. März 2012; abgerufen am 20. Februar 2016 (englisch).
- Nicole Kresge, Robert D. Simoni, Robert L. Hill: Succeeding in Science Despite the Odds; Studying Metabolism with NMR by Mildred Cohn. In: Journal of Biological Chemistry. Band 279, Nr. 53, 31. Dezember 2004, S. e12, PMID 15615732 (Online [abgerufen am 18. August 2010]).
- Alois Feusi: Winterthurer ETH-Professor Richard R. Ernst erhielt 1991 den Chemie-Nobelpreis für seinen Beitrag zur Entwicklung der Magnetresonanzspektroskopie. Neue Zürcher, 21. Mai 2020, abgerufen am 10. Oktober 2020.
- E. R. Andrew: Nuclear Magnetic Resonance. Cambridge at the University Press, 1958, S. 62.
- C. Reinhardt, T. Steinhauser: Formierung einer wissenschaftlich-technischen Gemeinschaft. NMR-Spektroskopie in der Bundesrepublik Deutschland. In: NTM Zeitschrift für Geschichte der Wissenschaften, Technik und Medizin. 16, 2008, S. 73–101, doi:10.1007/s00048-007-0280-z.
- A. Geiger, M. Holz: Automation and control in high power pulsed NMR. In: Journal of Physics E: Scientific Instruments. Band 13, Nr. 7, Januar 1980, S. 697, doi:10.1088/0022-3735/13/7/001.
- Nobelpreisvortrag von Wüthrich (PDF; 399 kB).
- R. Blachnik (Hrsg.): D'Ans, Lax: Taschenbuch für Chemiker und Physiker. 4. Auflage. Band 3: Elemente, anorganische Verbindungen und Materialien, Minerale. Springer-Verlag, Berlin 1998, ISBN 978-3-642-63755-1, S. 13, doi:10.1007/978-3-642-58842-6 (eingeschränkte Vorschau in der Google-Buchsuche).
- P. T. Callaghan: Principles of Nuclear Magnetic Resonance Microscopy. Clarendon Press, Oxford 1991.
- R. Neufeld, D. Stalke: Accurate Molecular Weight Determination of Small Molecules via DOSY-NMR by Using External Calibration Curves with Normalized Diffusion Coefficients. In: Chem. Sci. Nr. 6, 2015, S. 3354–3364, doi:10.1039/C5SC00670H.
- D. Li, I. Keresztes, R. Hopson, P. G. Williard: Characterization of Reactive Intermediates by Multinuclear Diffusion-Ordered NMR Spectroscopy (DOSY). In: Acc. Chem. Res. Nr. 42, 2008, S. 270–280, doi:10.1021/ar800127e.
- Matthias Findeisen, Stefan Berger: 50 and More Essential NMR Experiments – A Detailed Guide. Wiley-VCH, Weinheim, 4. Auflage, 2014, ISBN 978-3-527-33483-4.
- Timothy D.W. Claridge: High-Resolution NMR Techniques in Organic Chemistry. Elsevier, Amsterdam, 3. Auflage, 2016, S. 10, ISBN 978-0-08-099986-9.
- Gregory A. Manley, David Rovnyak: Observing Proton Exchange in Aqueous Ethanol with a 60 MHz FT-NMR Spectrometer. 2006.
- Andreas Maus, Christopher Hertlein, Kay Saalwächter: A Robust Proton NMR Method to Investigate Hard/Soft Ratios, Crystallinity, and Component Mobility in Polymers In: Macromolecular Chemistry and Physics. 207, 2006, S. 1150, doi:10.1002/macp.200600169.
- Hans-Otto Kalinowski: Der Schrecken der Weinpanscher. Quantitative Deuterium-NMR-Spektroskopie. In: Chemie in unserer Zeit. Band 22, Nr. 5, Oktober 1988, S. 162–171, doi:10.1002/ciuz.19880220503.
- Adolf Rapp, Alfred Markowetz: NMR-Spektroskopie in der Weinanalytik. In: Chemie in unserer Zeit. Band 27, Nr. 3, Juli 1993, S. 149–155, doi:10.1002/ciuz.19930270307.
- Bernd Wrackmeyer: NMR-Spektroskopie von Metallkernen in Lösung. In: Chemie in unserer Zeit. Band 28, Nr. 6, Dezember 1994, S. 309–320, doi:10.1002/ciuz.19940280610.
- Richard R. Ernst: Zürcher Beiträge zur 50-jährigen Entwicklung von Bruker. doi:10.1002/ange.201005067 (freier Volltext).