Impfstoffdesign
Das Impfstoffdesign (auch Impfstoffentwicklung) beschreibt Verfahren zur gezielten Anpassung von Impfstoffen. Das Impfstoffdesign ist eine Form des rationalen Designs und verwendet teilweise auch Methoden des Proteindesigns und des Vektordesigns.
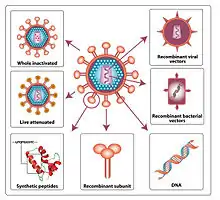
Eigenschaften
Mit einer Impfung soll ein längerfristiger Schutz vor einer Erkrankung und idealerweise eine Minderung der Weitergabe (Transmission) erzielt werden. Die adaptiven Teile des Immunsystems, die eine Form eines immunologischen Gedächtnisses ausbilden, sind bei Säugetieren Antikörper-produzierende B-Zellen, T-Helferzellen und zytotoxische T-Zellen. Für ein Impfstoffdesign müssen zuerst die in einem Impfstoff einzusetzenden Antigene identifiziert werden. Auf einem Antigen befinden sich meistens mehrere Epitope, an die Teile der adaptiven Immunantwort binden können.
Das Impfstoffdesign besteht als evolutive Methode aus einer Identifikation wirksamer Epitope anhand zuvor festgelegter Kriterien (engl. scoring ‚Punkte-Bewertung‘), gefolgt von Methoden zur Optimierung der Immunantwort. Im Gegensatz zur Gentherapie mit viralen Vektoren ist bei einer immunogenen Impfung mit viralen Vektoren ein längerfristiger Verbleib des Vektors im Impfling unerwünscht, da dies zur Ausbildung einer Immuntoleranz führen kann. Impfstoffe können immunogene (z. B. Impfstoffe gegen Pathogene oder Krebsimpfstoffe) oder tolerogene Wirkungen (Hyposensibilisierung, z. B. Glatirameracetat) erzeugen.[1] Entwicklungsparameter umfassen die Identifikation der zu verwendenden Antigene, die Applikationsform, Korrelate eines Impfschutzes, Tiermodelle, Skalierbarkeit, Produktionskapazitäten, das Zielprofil des Endprodukts, Vorhersage der Epidemiologie und die zu impfende Population.[2]
Bei attenuierten Viren, viralen Vektoren, DNA- und RNA-Impfstoffen wird der Impfstoff von den Zellen des Impflings hergestellt, mit nativer Konformation und korrekten posttranslationalen Modifikationen, im Gegensatz zu rekombinanten Proteinen, die in Zellkulturen aus anderen Klassen erzeugt wurden. Innerhalb der Zelle erfolgt eine Zerlegung der Antigene im Proteasom, eine Bindung der Fragmente (Peptide) an den Antigenpeptid-Transporter, ein Import des Peptids in das endoplasmatische Retikulum, eine Bindung des Peptids an MHCI und eine Exozytose des Peptid-MHCI-Komplexes an die Zelloberfläche. Dort werden die Peptid-MHCI-Komplexe den Immunzellen präsentiert und führen zu einer Aktivierung der zellulären Immunantwort.[3][4][5] Antigene außerhalb der Zelle werden per Endozytose in Endosomen aufgenommen, durch Peptidasen in Peptide zerlegt und dann von MHCII gebunden. Diese Komplexe werden per Exozytose an die Zelloberfläche sezerniert und ebenfalls den Immunzellen präsentiert. Daneben werden Membranproteine direkt auf der Zelloberfläche präsentiert, wo sie für B-Zellen zugänglich sind.
Identifikation
Die Epitope können durch zwei verschiedene Strategien charakterisiert werden. Die Epitope können aufgrund empirischer Kenntnisse über die Immunogenität bzw. die Korrelate des Impfschutzes gegen eine Infektion anhand einer vorhergehenden Bestimmung der Immunantwort gegen ein gezieltes Epitop gewählt werden. Alternativ können die Epitope aufgrund der Identifikation der Epitope durch eine Epitopkartierung mit Hilfe von Immunseren und Gedächtniszellen von Rekonvaleszenten ausgewählt werden.
Bewertungen
Verschiedene Ergebnisse können als maßgeblich ausgewählt werden, z. B. Induktion eines Titers, Wirksamkeit in einem Neutralisationstest,[6] ELISA oder ELISPOT oder durch einen Tierversuch nach Immunisierung und anschließender Infektion (Belastungsversuch, engl. challenge experiment) mit einer Erfassung des Pathogenitätsindexes und der Letalität. Hierbei können zur Minderung der Anzahl der Versuchsansätze die in-vitro-Methoden als begrenzte Vorschau verwendet werden. Die Überprüfung von Immunogenen erfolgt jedoch anschließend über einen Belastungsversuch, sofern ein Tiermodell der Infektion vorhanden ist. Gelegentlich wird auch ein Hämagglutinationshemmtest oder verschiedene Methoden zur Bestimmung der Zellviabilität antigenbeladener Opferzellen nach Zugabe von Immunzellen verwendet.
Neben den Kriterien zur Wirksamkeit (engl. efficacy) und Effizienz (engl. efficiency) des Impfstoffkandidaten werden auch die Wirksamkeit gegen mehrere Stämme eines Erregers, die Immundominanz, der Vektortyp, die Verabreichungsform, die Biologische Halbwertszeit, die Lagerstabilität, die Kosten, eine Erweiterbarkeit der Produktionskapazität im Epidemiefall, die Depotwirkung, die Dosis, eine Immunmodulation, der protektive Quotient und unerwünschte Arzneimittelwirkungen in eine Bewertung miteinbezogen. Als Ziele der Wirksamkeit können verschiedene Endpunkte herangezogen werden, wie eine Abwesenheit einer Infektion bei Geimpften (sterilisierende Immunität), eine Abwesenheit von Krankheit oder eine Abwesenheit von schweren Krankheitsverläufen. Je schwieriger ein Endpunkt zu erreichen ist, desto geringer ist die Wirksamkeit. Das bedeutet, dass derselbe Impfstoff höhere Werte der Wirksamkeit erreicht, wenn niedrigere Erfolgskriterien verlangt sind.
Korrelate eines Impfschutzes[7]
| Impfstoff | Typ | Testverfahren | Schutz ab |
|---|---|---|---|
| Hepatitis-A-Impfstoff | Inaktiviert | ELISA | 10 mIU/mL |
| Hepatitis-B-Impfstoff | HBsAg | ELISA | 10 mIU/mL |
| HPV-Impfstoff | Virus-like particle | ELISA | undefiniert |
| Influenzaimpfstoff | Gespalten oder attenuiert | HAI | 1:40 Titer |
| Japanische-Enzephalitis-Impfstoff | Inaktiviert oder attenuiert | Neutralisation | 1:10 Titer |
| Masernimpfstoff | Attenuiert | Neutralisation | 120–200 mIU/mL |
| Mumpsimpfstoff | Attenuiert | Neutralisation? | undefiniert |
| Polioimpfstoff | Attenuiert oder inaktiviert | Neutralisation | 1:4 bis 1:8 Titer |
| Tetanusimpfstoff | Inaktiviert | Neutralisation | 0.01 IU/mL |
| Tollwutimpfstoff | Inaktiviert | Neutralisation | 0.5 IU/mL |
| Rotavirus-Impfstoff | Attenuiert | Serum IgA | undefiniert |
| Rötelnimpfstoff | Attenuiert | Immunpräzipitation | 10–15 mIU/mL |
| Pockenimpfstoff | Attenuiert | Neutralisation | 1:20 bis 1:32 Titer |
| Varicellaimpfstoff | Attenuiert | FAMA, gpELISA, T-Zell-Proliferation | 1:64 Titer, 5 IU/mL, undefiniert |
| FSME-Impfung | Inaktiviert | Neutralisation | 125 IU/mL |
| Gelbfieberimpfstoff | Attenuiert | Neutralisation | 0.7 LNI |
Optimierungen
Zur Verstärkung einer Immunantwort gibt es verschiedene Strategien wie beispielsweise Wiederholungsimpfungen oder Dosiserhöhungen. Beim Prime-Boost-Verfahren werden mehrere Einzelkomponenten in definierter zeitlicher Abfolge gegeben, um eine additive oder synergistische Wirkung auf das Immunsystem zu erzielen. Man unterscheidet homologe (gleiche Start- und Wiederholungsvakzine) und heterologe (unterschiedliche Start- und Wiederholungvakzine) Prime-Boost-Schemata.[8] Auch eine Zugabe von Adjuvanzien kann zu einer Steigerung der Immunantwort führen.[9][10] Der Applikationsort hat einen Einfluss auf den entstehenden Antikörper-Subtyp: im Muskel werden eher IgG gebildet, die systemisch vor Erkrankung schützen, während in den Schleimhäuten eher IgA gebildet werden, welche an den Eintrittspforten vor Ansteckung schützen und so die Transmission mindern.
Weiterhin beschrieben sind die folgende Strategien bzw. Techniken. Zur Minderung der Effekte der Immunevasion und einem möglicherweise daraus folgenden Impfdurchbruch (insbesondere bei RNA-Viren und Bakterien) werden gelegentlich Konsensussequenzen eines Epitops aus verschiedenen Stämmen eines Pathogens oder Mischungen von Antigenen verschiedener Stämme (z. B. beim Influenzaimpfstoff) verwendet, um einen begrenzten Schutz gegen mehrere Stämme zu vermitteln.[11] Die Verwendung eines konservierten Epitops kann den Impfschutz auf mehrere Stämme ausdehnen.[12][13] Wiederholungen von Epitopen extrazellulärer Antigene können als Thymus-unabhängige Antigene eine humorale Immunantwort verstärken. Durch Aktivierung antigenpräsentierender Zellen kann die Impfantwort gesteigert werden.[14] Bei intrazellulären Pathogenen kann die Immunreaktion in Richtung zytotoxischer T-Zellen gelenkt werden, weil die humorale Immunantwort nicht innerhalb der Zelle wirken kann.[3] Retard-Formulierungen können die Antigenpräsentation zeitlich verlängern und so die Immunantwort steigern.[15] Durch virusartige Partikel und Virosomen kann eine gelegentlich schwache Immunogenität gereinigter Antigene (wie Untereinheitenimpfstoffe) teilweise kompensiert werden.[16] Durch einen geeigneten Vektor kann die Immunantwort gesteigert werden.[17] Bei genetischen Impfstoffen wie DNA-Impfstoffen (z. B. per Injektion oder per Genkanone), RNA-Impfstoffen oder viralen Vektoren werden die Antigene intrazellulär im Impfling hergestellt,[18] letztere können aufgrund einer Vektorimmunität nur einmal wirksam pro Geimpften eingesetzt werden. Daher wurden bei viralen Vektoren verschiedene Prime-Boost-Strategien zur Verstärkung der Immunantwort durch mehrfache Impfungen bei gleichzeitiger Umgehung einer Vektorimmunität entwickelt, bei der verschiedene Vektoren, jedoch mit dem gleichen Antigen, für die einzelnen Impfungen verwendet werden.
Geschichte
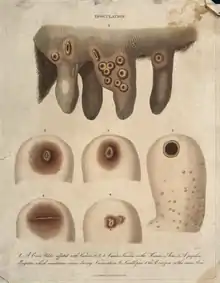
Die ersten Impfstoffe von Edward Jenner bestanden aus Pathogenen anderer Arten (z. B. Kuhpocken), die Humanpathogenen ausreichend ähnelten, um eine Immunantwort bei geringerer Erkrankung auszulösen. Neben der Verwendung von Pathogenen anderer Arten kamen in Folge weitere Methoden hinzu. Virale Impfstoffe der nächsten Generation bestanden aus inaktivierten (z. B. der Polioimpfstoff von Jonas Salk),[19] gespaltenen (z. B. der Influenzaimpfstoff) oder attenuierten Virionen, wie der 17D-Gelbfieberimpfstoff von Max Theiler,[20] der Impfstoff gegen das Poliovirus von Albert Sabin oder das Modified Vaccinia Ankara von Anton Mayr.[21][22]
Aus den Spaltimpfstoffen gingen die gereinigten Antigene (auch Untereinheiten-Impfstoffe, engl. subunit vaccines) wie das HBsAg des Hepatitis-B-Virus im Jahr 1981,[23] Konjugatimpfstoffe wie gegen Haemophilus influenzae im Jahr 1983 und auch die synthetisch erzeugten Peptidimpfstoffe hervor,[24][25] die durch eine Proteinreinigung bzw. im letzten Fall durch eine Peptidsynthese weniger Nebenwirkungen durch Kontaminationen erzeugten, weniger bzw. im letzten Fall kein Risiko einer Erkrankung besaßen und bei denen sich die Dosis leichter einstellen ließ, die jedoch auch oftmals weniger wirksam in Hinblick auf den Impfschutz waren. Diese Impfstoffe wirkten (mit Ausnahme der attenuierten Pathogene) vor allem außerhalb der Zelle auf die humorale Immunantwort, da nur eine geringe Aufnahme in Zellen und nur eine geringe anschließende Präsentation der Epitope an MHCI für eine zelluläre Immunantwort erfolgte. Daher wurden in den folgenden Jahren verstärkt genetische Impfstoffe entwickelt. Der erste für den Menschen zugelassene virale Vektorimpfstoff war ein Ebola-Impfstoff. Die ersten zugelassenen RNA- und DNA-Impfstoffe waren 2020 beziehungsweise 2021 SARS-CoV-Impfstoffe.
Markerimpfstoffe
Bei Markerimpfstoffen (synonym DIVA-Impfstoffe, von englisch Differentiating Infected from Vaccinated Animals ‚Unterscheidung von infizierten und geimpften Tieren‘) fehlt ein Epitop, wodurch Geimpfte und Erkrankte unterschieden werden können. Bei Geimpften fehlt die Immunreaktion gegen dieses Epitop.
Literatur
- C. Janeway et al.: Immunobiology (= Spektrum-Lehrbuch.). 5. Auflage, Spektrum, Akademischer Verlag, Berlin/ Heidelberg 2002, ISBN 3-8274-1079-7 ((online)).
Einzelnachweise
- P. Moingeon, V. Lombardi, N. Saint-Lu, S. Tourdot, V. Bodo, L. Mascarell: Adjuvants and vector systems for allergy vaccines. In: Immunology and Allergy Clinics of North America. 2011, Band 31, Nr. 2, S. 407–419, xii, doi:10.1016/j.iac.2011.03.001, PMID 21530828.
- E. Prompetchara, C. Ketloy, T. Palaga: Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. In: Asian Pacific journal of allergy and immunology. März 2020, Band 38, Nr. 1, S. 1–9, doi:10.12932/AP-200220-0772, PMID 32105090.
- R. A. Koup, D. C. Douek: Vaccine design for CD8 T lymphocyte responses. In: Cold Spring Harbor Perspectives in Medicine. 2011, Band 1, Nr. 1, S. a007252, doi:10.1101/cshperspect.a007252, PMID 22229122, PMC 3234456 (freier Volltext).
- G. Ada, I. Ramshaw: DNA vaccination. In: Expert Opinion on Emerging Drugs. 2003, Band 8, Nr. 1, S. 27–35, PMID 14610909.
- J. C. Nolz, J. T. Harty: Strategies and implications for prime-boost vaccination to generate memory CD8 T cells. In: Advances in Experimental Medicine and Biolog. 2011, Band 780, S. 69–83, doi:10.1007/978-1-4419-5632-3_7, PMID 21842366.
- M. Bonsignori, S. M. Alam, H. X. Liao, L. Verkoczy, G. D. Tomaras, B. F. Haynes, M. A. Moody: HIV-1 antibodies from infection and vaccination: insights for guiding vaccine design. In: Trends in Microbiology. 2012, Band 20, Nr. 11, S. 532–539, doi:10.1016/j.tim.2012.08.011, PMID 22981828; PMC 3757512 (freier Volltext).
- I. J. Amanna, M. K. Slifka: Contributions of humoral and cellular immunity to vaccine-induced protection in humans. In: Virology. 2011, Band 411, Heft 2, S. 206–215, doi:10.1016/j.virol.2010.12.016, PMID 21216425, PMC 3238379 (freier Volltext).
- Shan Lu: Heterologous Prime-Boost Vaccination. In: Current Opinion in Immunology. Juni 2009, Band 21, Nr. 3, S. 346–351, doi:10.1016/j.coi.2009.05.016.
- M. G. Tovey, C. Lallemand: Adjuvant activity of cytokines. In: Methods in Molecular Biology. 2010, Band 626, S. 287–309, doi:10.1007/978-1-60761-585-9_19, PMID 20099135.
- G. C. Bowick, A. J. McAuley: Vaccine and adjuvant design for emerging viruses: mutations, deletions, segments and signaling. In: Bioengineered Bugs. 2011, Band 2, Nr. 3, S. 129–135, PMID 21637006, PMC 3225654 (freier Volltext).
- N. Miller: Recent progress in dengue vaccine research and development. In: Current Opinion in Molecular Therapeutics. 2010, Band 12, Nr. 1, S. 31–38, PMID 20140814.
- S. M. Kang, J. M. Song, R. W. Compans: Novel vaccines against influenza viruses. In: Virus Research. 2011, Band 162, Nr. 1–2, S. 31–38, doi:10.1016/j.virusres.2011.09.037, PMID 21968298, PMC 3401575 (freier Volltext).
- K. Kaur, M. Sullivan, P. C. Wilson: Targeting B cell responses in universal influenza vaccine design. In: Trends in Immunology. 2011, Band 32, Nr. 11, S. 524–531, doi:10.1016/j.it.2011.08.007, PMID 21940217, PMC 3212832 (freier Volltext).
- R. M. Roy, B. S. Klein: Dendritic cells in antifungal immunity and vaccine design. In: Cell Host & Microbe. 2012, Band 11, Nr. 5, S. 436–446, doi:10.1016/j.chom.2012.04.005, PMID 22607797, PMC 3401965 (freier Volltext).
- L. Han, K. Peng, L. Y. Qiu, M. Li, J. H. Ruan, L. L. He, Z. X. Yuan: Hitchhiking on Controlled-Release Drug Delivery Systems: Opportunities and Challenges for Cancer Vaccines. In: Frontiers in pharmacology. Band 12, 2021, S. 679602, doi:10.3389/fphar.2021.679602, PMID 34040536, PMC 8141731 (freier Volltext).
- A. Zeltins: Construction and characterization of virus-like particles: a review. In: Molecular Biotechnology. 2013, Band 53, Nr. 1, S. 92–107, doi:10.1007/s12033-012-9598-4, PMID 23001867.
- J. C. Small, H. C. Ertl: Viruses - from pathogens to vaccine carriers. In: Current Opinion in Virology. 2011, Band 1, Nr. 4, S. 241–245, doi:10.1016/j.coviro.2011.07.009, PMID 22003377, PMC 3190199 (freier Volltext).
- F. Saade, N. Petrovsky: Technologies for enhanced efficacy of DNA vaccines. In: Expert Review of Vaccines. 2012, Band 11, Nr. 2, S. 189–209, doi:10.1586/erv.11.188, PMID 22309668, PMC 3293989 (freier Volltext).
- J. E. Salk: Studies in human subjects on active immunization against poliomyelitis. I. A preliminary report of experiments in progress. In: Journal of the American Medical Association. 1953, Band 151, Nr. 13, S. 1081–1098, PMID 13034436.
- M. Theiler, H. H. Smith: The effect of prolonged cultivation in vitro upon the pathogenicity of Yellow Fever Virus. In: Journal of Experimental Medicine. 1937, Band 65, Nr. 6, S. 767–786, PMID 19870633, PMC 2133530 (freier Volltext).
- A. B. Sabin: Present status of attenuated live virus poliomyelitis vaccine. In: Bulletin of the New York Academy of Medicine 1957, Band 33, Nr. 1, S. 17–39, PMID 13383294, PMC 1806054 (freier Volltext).
- A. Mayr, V. Hochstein-Mintzel, H. Stickl: Abstammung, Eigenschaften und Verwendung des attenuierten Vaccinia-Stammes MVA. In: Infection. 1975, Band 3, S. 6–14.
- R. H. Purcell, J. L. Gerin: Hepatitis B subunit vaccine: a preliminary report of safety and efficacy tests in chimpanzees. In: American Journal of the Medical Sciences. 1975, Band 270, Nr. 2, S. 395–399, PMID 828832.
- R. Schneerson, O. Barrera, A. Sutton, J. B. Robbins: Preparation, characterization, and immunogenicity of Haemophilus influenzae type b polysaccharide-protein conjugates. In: Journal of Experimental Medicine. 1980, Band 152, Nr. 2, S. 361–376, PMID 6967514, PMC 2185954 (freier Volltext).
- A. Patronov, I. Doytchinova: T-cell epitope vaccine design by immunoinformatics. In: Open Biology. 2013, Band 3, Nr. 1, S. 120139, doi:10.1098/rsob.120139, PMID 23303307, PMC 3603454 (freier Volltext).