Hämostase
Die Hämostase (zusammengesetzt aus altgriechisch αἷμα haíma, deutsch ‚Blut‘, und Stase von στάσις stasis, deutsch ‚Stauung‘, ‚Stillung‘, ‚Stockung‘, ‚Stillstand‘) ist ein lebenswichtiger Prozess, der die bei Verletzungen der Blutgefäße entstehenden Blutungen zum Stehen bringt. Dadurch wird der übermäßige Austritt von Blut aus dem Blutkreislauf verhindert und die Voraussetzung für eine Wundheilung geschaffen. Die Hämostase muss im Fall einer Verletzung hinreichend schnell einsetzen, um größeren Blutverlust zu vermeiden. Sie muss auf den Bereich der Verletzung beschränkt bleiben und darf nicht fälschlicherweise durch andere Ereignisse wie Entzündungen oder Infektionen ausgelöst werden.
Die Hämostase lässt sich in zwei Teilvorgänge aufteilen, die jedoch miteinander in Wechselwirkung stehen. Bei der primären (auch: zellulären) Hämostase, der (physiologischen) Blutstillung, sind die Blutplättchen (Thrombozyten), die Wandzellen des betroffenen Blutgefäßes (Endothel und glatte Muskelzellen), sowie Gewebe außerhalb des Gefäßes beteiligt. Vereinfacht dargestellt verengt sich das Gefäß zunächst, dann heften sich Blutplättchen an das Leck, verkleben untereinander und stellen so den ersten Wundverschluss her. Bei der sekundären (auch: plasmatischen) Hämostase, der Blutgerinnung, wird dieser noch lose Verschluss durch die Bildung von Fibrin-Fäden verstärkt. Hierbei spielt die Aktivierung von etwa einem Dutzend im Blutplasma enthaltenen Gerinnungsfaktoren eine wichtige Rolle. Ein genetischer Defekt von Gerinnungsfaktoren kann zu Krankheiten wie der Hämophilie (Bluterkrankheit) führen. Die einsetzende Wundheilung wird durch Wachstumsfaktoren initiiert, die von Thrombozyten und Endothelzellen abgegeben werden. Am Ende der Wundheilung wird das Fibrin durch das fibrinolytische System des Blutplasmas aufgelöst.
Die Hämostase wird nicht selten auch mit ihren beiden Anteilen, der primären und der sekundären Hämostase, als Blutstillung bezeichnet.[1]
Unter Hyperkoagulabilität versteht man die erhöhte Gerinnbarkeit des Blutes. Die Hämostaseologie ist die Lehre von der Hämostase. Unter einer Koagulopathie oder einer Blutgerinnungsstörung versteht man eine krankhaft veränderte Hämostase.
Dieser Artikel beschreibt die Hämostase beim Menschen. Die Aussagen treffen überwiegend auch auf andere Säuger zu, aber nur eingeschränkt auf andere Tierklassen.
Physiologische Vorgänge nach einer Gefäßverletzung

Nach Verletzung kleinerer Gefäße kommt eine Blutung üblicherweise zügig zum Stehen. Die dafür verantwortliche Hämostase kann als Abfolge der folgenden Prozesse betrachtet werden. Diese Unterteilung dient in erster Linie dem einfacheren Verständnis. Zwischen den Prozessen bestehen enge funktionelle und zeitliche Beziehungen, eine scharfe Abgrenzung ist nicht möglich.
- Spontane arterielle Hämostase
- Schlagadern (Arterien) vom muskulären Typ haben die Eigenschaft, sich nach einer Querdurchtrennung von selbst „einzukrempeln“. Diese Eigenschaft liegt im Wandbau der Schlagadern begründet: Die elastische Innenhaut der Schlagader (Membrana elastica interna) zieht sich nach Durchtrennung stärker zusammen als die übrigen Wandschichten. Dadurch wird der freie Rand der durchtrennten Schlagader in das Innere des Gefäßes hineingezogen und sorgt so für einen sehr schnellen, provisorischen Verschluss.[3]
- Zelluläre Hämostase
- Sie besteht aus der Anheftung (Adhäsion) und Verklebung (Aggregation) von Thrombozyten, der Aktivierung weiterer Thrombozyten und Bildung eines verschließenden, weißen Thrombozytenthrombus. Außerdem wird durch die Ausschüttung von Substanzen eine Vasokonstriktion, also eine Gefäßverengung, ausgelöst. Dies verringert den Blutfluss und minimiert so den Blutverlust.
- Plasmatische Hämostase
- Bestandteile des Blutplasmas erzeugen ein Maschenwerk aus mechanisch stabilen Fibrinfäden, worin die zirkulierenden roten Blutkörperchen (Erythrozyten) hängen bleiben und sich schließlich ein roter Thrombus bildet, der sich schließlich verfestigt und zusammenzieht.
Zelluläre Hämostase
Das Blut eines Menschen enthält im Normalfall zwischen 150.000 und 400.000 Thrombozyten pro Mikroliter.[4] In der Zellmembran der Thrombozyten sind zahlreiche Glykoproteine und Rezeptoren vorhanden, die bei der zellulären Hämostase eine wichtige Rolle spielen.
Die innere Zellschicht von Blutgefäßen wird als Endothel bezeichnet. Diese ist innen mit einer Glykokalyx, einer Art Schleimschicht überzogen, für die Thrombozyten keine Rezeptoren besitzen. Unter anderem aus diesem Grund bleiben Thrombozyten in unverletzten Gefäßen inaktiv und können sich nicht an die Gefäßwand anlagern. Verschiedene Faktoren wirken einer Aktivierung ebenfalls entgegen, beispielsweise Prostacyclin und Stickstoffmonoxid sowie Heparin, das unter anderem von Mastzellen gebildet wird und dessen hemmende Wirkung auf die Hämostase therapeutisch genutzt werden kann.
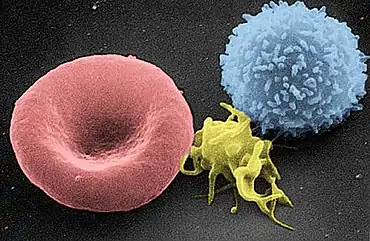
Thrombozytenadhäsion und Aktivierung
Wenn ein Gefäß verletzt wird, kommt das Blut mit dem umliegenden Bindegewebe in Berührung, unter anderem mit Kollagenfasern. Kollagen ist ein Strukturprotein, das nahezu überall im Extrazellularraum vorhanden ist. Die Thrombozyten haften zuerst an diesen Fasern (Thrombozytenadhäsion), was zur Ausbildung einer dünnen Bedeckung der Wunde führt.[5] Die Adhäsion (Anhaftung) wird durch den Von-Willebrand-Faktor (vWF) vermittelt, ein lösliches Blutprotein, welches von Endothelzellen und Megakaryozyten gebildet wird. Er stellt zusammen mit Fibronektin und Laminin eine Verbindung zwischen Kollagenfasern und einem Rezeptor auf den Thrombozyten (GP Ib/V/IX) her. Ein Defekt des Von-Willebrand-Faktors führt zum Willebrand-Jürgens-Syndrom.
Durch die Adhäsion wird die Thrombozytenaktivierung ausgelöst: Sie setzen aus sogenannten „elektronendichten Granula“ Calcium-Ionen, ADP, Serotonin, Thromboxan A2 und weitere Stoffe frei. Dadurch werden weitere Thrombozyten angelockt (Chemotaxis). Thromboxan A2 trägt außerdem maßgeblich zur Verengung des Blutgefäßes bei, die einem hohen Blutdurchfluss entgegenwirkt. Auch der Inhalt der sogenannten „α-Granula“ der Thrombozyten wird ausgeschüttet: Gerinnungsfaktoren (Faktor V, Faktor VIII), Klebstoffe (vWF, Fibronektin, Thrombospondin) und Wachstumsfaktoren.[6] Durch Aktivierung verschiedener Stoffwechselwege werden vermehrt Substanzen wie Thromboxan A2 und der PAF (Platelet Activating Factor, plättchenaktivierender Faktor) gebildet. Einige dieser Stoffe induzieren die plasmatische Gerinnung.
Thrombozytenaggregation

Die Zusammenlagerung (Aggregation) der aktivierten Thrombozyten wird gefördert durch eine Umorganisation des Zytoskeletts, die eine Vergrößerung der Zelloberfläche um ein Mehrfaches bewirkt. Während die Thrombozyten inaktiv Linsenform haben, nehmen sie im aktiven Zustand Kugelform an und tragen dabei lange Pseudopodien (Schein-Füßchen), mit deren Hilfe sie sich untereinander einhaken können – die Thrombozyten werden „stachelig“ und „klebrig“. Die aggregierten Thrombozyten bilden schließlich einen Thrombozyten-Pfropf, der als weißer Thrombus bezeichnet wird. Damit endet die zelluläre Hämostase. Normalerweise dauert der Prozess ein bis vier Minuten, diese Dauer wird als Blutungszeit bezeichnet.
Der weiße Thrombus ist nicht allzu stabil und kann weggeschwemmt werden. Einen festeren Verschluss bildet die plasmatische Hämostase.
Plasmatische Hämostase
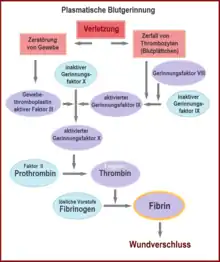
Durch die plasmatische Hämostase bildet sich ein Maschenwerk aus mechanisch stabilem Fibrin, in das neben Thrombozyten auch rote Blutkörperchen (Erythrozyten) eingefangen werden, der daher als „roter Thrombus“ bezeichnet wird.
Aktivierte Thrombozyten haben auf der Zellmembran einen Rezeptorkomplex (Glycoprotein IIb/IIIa), an welchem Fibrinogen aus dem Plasma und die aus den aktivierten Thrombozyten freigesetzten Haftstoffe (Fibronektin, Thrombospondin) binden. Rückkopplungsmechanismen der ausgeschütteten Stoffe führen schließlich zu einer irreversiblen Aggregation, bei der die Zellmembranen der Thrombozyten miteinander verschmelzen.[7]
Diese sekundäre Blutstillung, die Blutgerinnung, wird auch als Gerinnungskaskade bezeichnet. Sie wird in drei Phasen unterteilt: Aktivierungs-, Koagulations- und Retraktionsphase.
Aktivierungsphase
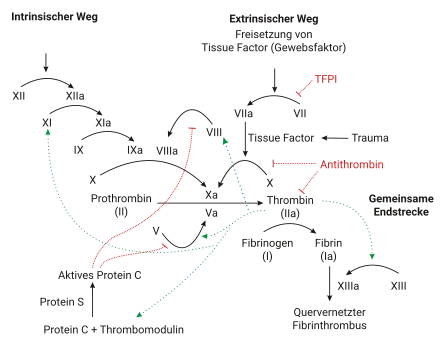
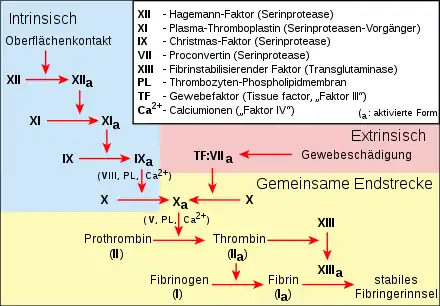
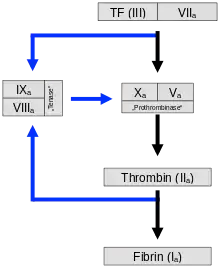
Durch den Kontakt von Thrombozyten mit negativ geladenen Oberflächen wie zum Beispiel Glas werden die Faktoren XII und XI aktiviert, die eine Gerinnungskaskade in Gang setzen (intrinsisches System, siehe Abbildung). Wird der Faktor XII bei einem Individuum nicht gebildet, hat dies keine bedeutende Störung der Gerinnung zur Folge, im Gegensatz zum Mangel der Faktoren VIII, IX und XI,[8] der zur Hämophilie A, B und C führt.
Der normale physiologische Ablauf[9] (extrinsisches System oder exogener Mechanismus) wird durch Kontakt von Blut mit Gewebethromboplastin aus verletztem subendothelialen Gewebe initiiert. Gewebefaktor (auch Tissue Factor (TF), Gewebethromboplastin oder Faktor III) ist ein Membranprotein, welches beispielsweise in der Adventitia von Blutgefäßen vorkommt – von Endothelzellen wird es nur nach Aktivierung freigesetzt. Es bildet einen Komplex mit Faktor VII, der in seine aktive Form überführt wird. Dadurch wird etwas Thrombin gebildet, der Prozess wird aber relativ schnell durch den TFPI (Tissue Factor Pathway Inhibitor) gehemmt. Wenn genug Thrombin gebildet wurde, wird ein sogenannter Aktivatorkomplex der Faktoren IX und VIII aktiviert (siehe IXa und VIIIa in Abbildung). Dieser Komplex aktiviert wiederum Faktor X.
Das Fehlen der Faktoren VIII oder IX führt zur Hämophilie, der Bluterkrankheit: Die Kaskade wird unterbrochen und die Verstärkung der Gerinnung bleibt aus. Die Patienten können an kleinsten inneren Verletzungen verbluten.
Bei beiden Mechanismen – intrinsischer und extrinsischer Weg – wird schließlich Faktor X zu Faktor Xa aktiviert. Dieser wiederum spaltet Prothrombin (Faktor II), es entsteht Thrombin (Faktor IIa). Diese Reaktion auf der Thrombozyten-Membran findet nur in Anwesenheit von Calcium statt und wird durch positive Rückkopplung mit dem Komplex der Faktoren VIII und IX stark beschleunigt. Mit der Bildung von enzymatisch aktivem Thrombin endet die Aktivierungsphase.
Phasen der Koagulation und Retraktion
Das enzymatisch aktive Thrombin ist für die Polymerisation von Fibrin und damit die Bildung des roten Thrombus verantwortlich: In der Koagulationsphase spaltet es aus der inaktiven Vorstufe Fibrinogen (Faktor I) niedermolekulare Einheiten (Monomere) ab, welche sich nichtkovalent zum polymeren Fibrin zusammenlagern. Durch Wirkung des Faktors XIII werden zwischen den Monomeren schließlich kovalente Bindungen geknüpft und der Thrombus wird stabilisiert. Das Fibrin vernetzt die schon aneinandergelagerten Thrombozyten und festigt damit den Wundverschluss. In das Netz werden rote Blutkörperchen eingefangen, ein sogenannter roter Thrombus bildet sich. Das Thrombin bewirkt weiterhin eine Kontraktion des Aktin-Myosin-Skeletts innerhalb der Thrombozyten: Die sich kontrahierenden Thrombozyten ziehen am Fibrinnetz und somit die Wundränder zusammen und verschließen die Wunde mechanisch. Durch das Zusammenziehen – unterstützt durch den PDGF (platelet-derived growth factor) – wird außerdem das Eindringen von Bindegewebszellen gefördert: die Wundheilung beginnt.
Neues zellbasiertes Modell der Gerinnung[10]
Das klassische Modell der Gerinnung unterscheidet eine intrinsische und extrinsische Aktivierung und beschreibt damit eine mehrstufige Abfolge der Aktivierung von Proteinen im zellfreien Plasma. Die klassischen Gerinnungstests aPTT und PT entsprechen dieser Vorstellung. Um die Blutgerinnung an beschädigten Blutgefäßen im Körper zu beschreiben, ist dieses Modell nicht geeignet, so dass sich 2001 ein zellbasiertes Modell der Gerinnung etablierte, welches drei überlappende Phasen beschreibt:
Initiation
Durch eine Gewebeverletzung kommen ansonsten außerhalb des Blutgefäßes befindliche Zellen unterhalb des Gefäßendothels in Kontakt mit dem Blutstrom. Die nun offen im Blutstrom liegenden Zellen tragen den Gewebsfaktor (III) auf ihrer Oberfläche (tissue-factor-(III)-bearing cell). Der Komplex aus Tissue-Faktor (III) und Proconvertin (VII) katalysiert nun die Aktivierung von Thrombokinase (X), welche vorerst nur geringe Mengen Thrombin (II) aktivieren kann. Diese geringe Menge an Thrombin (II) reicht jedoch aus, um Thrombozyten sowie die Gerinnungsfaktoren Proaccelerin (V) und Proconvertin(VII) zu aktivieren und damit die Amplifikation der Thrombinbildung anzustoßen.[11] Diese erste Phase findet auf kleinem Raum an der subendothelialen Verletzung statt.
Amplifikation
Das in der Initiationsphase aktivierte Proaccelerin (V) bildet mit der Thrombokinase (X) den Prothrombinasekomplex (X,V), welcher verstärkt Thrombin (II) aktivieren kann. Zeitlich überlappend verläuft die Amplifikationsphase, bei der Thrombozyten sich an subendotheliale Strukturen anheften (Adhäsion). Dies geschieht über GPVI-Rezeptoren, die an Kollagen binden sowie über den GPIb/IX-Rezeptor, der an Von-Willebrand-Faktor bindet. Der Von-Willebrand-Komplex gibt dabei den gebundenen Faktor VIII frei, der in aktiver Form an die Oberfläche der Thrombozyten bindet. Weiterhin schüttet der Thrombozyt seine inneren Vorräte aus, die unter anderem auch Proaccelerin (V) enthalten. Der Tissue-Faktor(III) und Proconvertin(VII) aktivieren nicht nur Thrombokinase (X), sondern auch Serinprotease (IX). Die ersten kleinen Mengen Thrombin (II) aktivieren nicht nur Fibrin (I), sondern auch den Faktor (VIII). Der aus VIII und IX gebildete Tenasekomplex aktiviert wiederum die Thrombokinase (X), so dass sich eine sich selbst verstärkende Schleife bildet. Dieser wichtige Schritt wird auch als Josso-Loop bezeichnet. Nun befinden sich auf der Oberfläche der subendothelialen Verletzung sowie auf der Oberfläche des dort anheftenden Thrombozyten zahlreiche Gerinnungsfaktoren in hoher Konzentration und sind geschützt vor den antikoagulatorischen Proteinen im freien Blut.
Propagation
Die auf der Thrombozytenoberfläche angehäuften Faktoren bilden Tenasekomplexe (VIII, IX), die die Bildung des Prothrombinasekomplex (X, V) unterstützen. Durch die Thrombokinase (X) werden nun große Mengen Thrombin (II) aktiviert (Thrombin-Burst). Thrombin (II) bildet schließlich die Fibrinnetze, in die sich Thrombozyten mit ihren GPIIb/IIIa-Rezeptoren einbinden. Faktor XIII stabilisiert diese Netze durch zusätzliche Fibrin-Querverbindungen.
Kontrolle vor ungewollter Ausbreitung des Gerinnsels
Um zu verhindern, dass sich außerhalb der Endothelverletzung ein Gerinnsel bildet (Thrombose), verfügt das Endothel und das frei fließende Blut über verschiedene Mechanismen: Sich lösendes Thrombin (II) wird im Blutstrom zügig durch das antikoagulatorische Protein Antithrombin deaktiviert. Sich lösende Thrombokinase (X) und Proconvertin (VII) wird durch TFPI gebunden. Auf der Oberfläche des Endothel befindet sich Thrombomodulin (TM), das Trombin (II) bindet, so dass dieses kein Fibrin (I) mehr bilden kann. Gleichzeitig wird die Bildung von Protein C (APC) durch das gebundene Thrombin (II) vertausendfacht, so dass Thrombin nun antikoagulatorisch wirkt. Protein C (APC) bildet dann mit Protein S einen Komplex, der die Gerinnungsfaktoren V und VIII deaktiviert. Weiterhin verfügt das Endothel über membranständige ADPasen, die ADP abbauen und damit die Thrombozytenfunktion herunterregeln.
Übergang zur Wundheilung
Nach der plasmatischen Hämostase erfolgt die Wundheilung, indem Bindegewebe bildende Zellen (Fibroblasten) in den Thrombus einwachsen und ihn bindegewebig umbauen. Dabei sterben beschädigte Zellen ab und werden abgebaut. Für den Abbau der Thromben ist vor allem ein Protein namens Plasmin zuständig, das ebenfalls durch kompliziert regulierte Mechanismen aus einer inaktiven Vorstufe (Plasminogen) gebildet wird. Plasmin löst die kovalenten Bindungen zwischen den Fibrin-Strängen auf und damit das Netz, das den Thrombus festhält.
Zwischen den Systemen der Blutgerinnung und dem System der Fibrinolyse, welches den roten Thrombus im Gefäßsystem wieder auflöst, bestehen abgestimmte Gleichgewichte. Geringfügige Störungen dieser Gleichgewichte können zu schwerwiegenden Blutungen oder zur Bildung von Thromben an Orten führen, an denen keine Verletzung vorliegt (siehe auch Thrombose).
Übersicht über Gerinnungsfaktoren und Inhibitoren
Bis auf Calciumionen (Faktor IV) sind die Gerinnungsfaktoren Eiweiße (Proteine). Jedem Faktor ist eine römische Zahl zugewiesen. Ein kleines a hinter der Zahl bedeutet, dass er in der aktiven Form vorliegt. Aus historischen Gründen (siehe unter Forschungsgeschichte) ist die Zahl VI nicht (mehr) vergeben, der entsprechende Faktor ist identisch mit Va.
| Nummer | Name(n) | Funktionen | Mangelsyndrome |
|---|---|---|---|
| I | Fibrinogen | Vorläufermolekül zur Bildung des Fibrinnetzes. | Afibrinogenämie (angeboren oder bei Verbrauchskoagulopathie) |
| II | Prothrombin | Die aktive Form Thrombin (IIa) aktiviert die Faktoren I, V, VIII, XI und XIII. | Hypoprothrombinämie (angeboren, Vitamin-K-Mangel oder bei Verbrauchskoagulopathie) |
| III | Gewebefaktor, Gewebethromboplastin, Tissue factor (TF) | Als einziger nicht im Blut, sondern im subendothelialen Gewebe.
TF und VIIa bilden mit Ca2+ die extrinsische Tenase, die X aktiviert. |
|
| IV | Calcium | Viele Faktoren benötigen das Calcium-Kation Ca2+, um an die negativ geladenen Phospholipide der Plasmamembranen zu binden. | Calciummangel |
| V | Proaccelerin | Va und Xa bilden mit Ca2+ und Phospholipiden den Prothrombinasekomplex, der II aktiviert. | Parahämophilie (angeboren) |
| VI | entspricht Faktor Va | ||
| VII | Proconvertin | VIIa und TF bilden mit Ca2+ die extrinsische Tenase, die X aktiviert. | Hypoprokonvertinämie (angeboren, Vitamin-K-Mangel) |
| VIII | Antihämophiles Globulin A | VIIIa und IXa bilden mit Ca2+ und Phospholipiden die intrinsische Tenase, die X aktiviert. | Hämophilie A (angeboren, X-chromosomal rezessiv vererbt) |
| IX | Christmas-Faktor, Antihämophiles Globulin B | VIIIa und IXa bilden mit Ca2+ und Phospholipiden die intrinsische Tenase, die X aktiviert. | Hämophilie B (angeboren, X-chromosomal rezessiv vererbt) |
| X | Stuart-Prower-Faktor | Va und Xa bilden mit Ca2+ und Phospholipiden den Prothrombinasekomplex, der II aktiviert. | Faktor-X-Mangel (angeboren) |
| XI | Rosenthal-Faktor, Plasma Thromboplasmin Antecedent (PTA) | XIa aktiviert IX. | Hämophilie C (angeboren) oder PTA-Mangel bei Verbrauchskoagulopathie |
| XII | Hageman-Faktor | XIIa aktiviert XI. | Hageman-Syndrom führt eher zu Störungen der Fibrinolyse (angeboren oder bei Verbrauchskoagulopathie) |
| XIII | Fibrinstabilisierender Faktor | XIIIa wandelt Fibrinmonomere in vernetztes Fibrin um. | Faktor-XIII-Mangel |
Um eine Gerinnung in der Abwesenheit von Verletzungen zu vermeiden, enthält das Blutplasma verschiedene hemmende Substanzen (Inhibitoren). Proteaseinhibitoren hemmen die Bildung von Fibrin. Antithrombin hemmt mehrere Gerinnungsproteasen in der Aktivierungsphase und Koagulationsphase. Die inhibitorische Wirkung wird durch seinen Kofaktor, das Heparin, deutlich verstärkt. Heparin wird von Endothelzellen und Mastzellen gebildet. Thrombomodulin, das ebenfalls aus dem Endothel stammt, bindet an Thrombin und aktiviert Protein C, das nach Bindung an Protein S die Cofaktoren Va und VIIIa inaktiviert.
Forschungsgeschichte
Bis 1772 hielt man an dem Gedanken von Aristoteles fest, dass das Gerinnen des Blutes mit dem Gefrieren von Flüssigkeiten zu vergleichen ist.[13]
Die ersten Theorien der Hämostase deuteten geronnenes Blut vor dem Hintergrund der Humoralpathologie als „Schwarze Galle“. Ab dem 17. Jahrhundert wurde mit der Untersuchung der physiologischen Mechanismen begonnen.[14] 1772 zeigte William Hewson, dass im Blut eine Lymphe vorhanden ist, die für die Koagulation verantwortlich ist.[13] Im 18. Jahrhundert herrschte zwischenzeitlich wieder die Meinung, dass bei der Blutgerinnung die Bewegung des Blutes zum Erliegen kommt und ein Absetzen der suspendierten Teilchen dazu führt, dass der Eindruck der Blutgerinnung eintritt. 1821 gelang durch Jean Louis Prevost und Jean Baptiste André Dumas der Durchbruch in der Forschung: Die Gerinnung ist ein Zusammentreten von Blutkörperchen und Fibrin. Johannes Müller stellte fest, dass das Fibrin im Blut gelöst sein muss, weitere Erklärung konnte 1856 dann Rudolf Virchow liefern, indem er auf die Vorstufe des Fibrins, die er Fibrinogen nannte, stieß. 1830 bis 1859 führte Prosper Sylvain Denis de Commercy (1799–1863) mehrere Studien durch, in denen er unter anderem die Instabilität der Gerinnsel feststellte. Ihm gelang auch die Fällung von Serofibrin aus dem Plasma, welches er das Plasmin nannte.[13]
Alexander Schmidt (1831–1894) stellte 1876 eine Gerinnungstheorie auf, die auf miteinander in Wechselwirkung stehenden Proteinen basierte. Auch die Rolle des Calciums wurde von ihm beschrieben.[14] Lange wurde diskutiert, welche Stoffe zur Gerinnung wirklich nötig sind und ob die zelluläre oder die plasmatische Phase die bedeutendere sei.
Olof Hammarsten (1841–1932) und Léon Fredericq (1851–1939) zeigten 1875, dass Fibrinferment und Fibrinogen die einzigen Substanzen sind, die zur Blutstillung führen und es sich nicht um eine fibrinoplastische Substanz handelt. Schmidt forschte an diesem Ferment weiter und gab ihm den Namen Thrombin. Außerdem erstellte er die These, dass es Prothrombin im Plasma geben muss.[13]
Im Jahr 1904 beschrieb Paul Morawitz das System schon fast so, wie es heute bekannt ist. Er prägte den Begriff der plasmatischen Gerinnung und beschrieb die folgenden zwei Phasen


Die molekularen Mechanismen der Blutgerinnung wurden zum größten Teil im Laufe des 20. Jahrhunderts entdeckt. Ein erster Hinweis auf die Komplexität der Mechanismen der Blutgerinnung war die Entdeckung von Proaccelerin durch Paul Owren (1905–1990) im Jahre 1947, welches als Faktor V bezeichnet wurde. Die komplette Aminosäuresequenz wurde 1987 durch Jenny et al. veröffentlicht.[15] Owren vermutete bereits, dass dieser Faktor Accelerin produziert, das er als Faktor VI bezeichnete. Später stellt sich heraus, dass Faktor V die inaktive Vorstufe von Faktor VI ist. Deshalb wird Faktor VI nun als Faktor Va bezeichnet.
Faktor IX wurde 1952 auf Grund der Krankheit eines jungen Patienten mit Hämophilie B namens Stephen Christmas entdeckt, bei dem das Fehlen ebendieses Faktors die Krankheit auslöste. Er heißt deshalb Christmas-Faktor.[16] Viele der anderen Faktoren wurden ebenfalls in den 1950er Jahren entdeckt und häufig nach den Patienten benannt, in denen sie gefunden wurden. Details zu diesen Entdeckungen sind in den Artikeln der jeweiligen Faktoren beschrieben.
Erst in neuerer Zeit wurde entdeckt, dass der intrinsische Weg wohl keine physiologische Rolle spielt, das heißt, dass er in vitro, nicht aber in vivo beobachtet werden kann.[17]
Klinische Bedeutung
Medikamentöse Gerinnungshemmung
Vor, während und nach Operationen sowie bei Bettlägerigkeit aus anderer Ursache werden häufig vorübergehend gerinnungshemmende (fälschlicherweise oft als Blutverdünner bezeichnete) Medikamente zur Vermeidung von Thrombosen und Lungenembolien eingesetzt. Diese Vorgehensweise wird Thromboseprophylaxe genannt.
Häufigster Grund für eine längerfristige therapeutische Antikoagulation ist heutzutage das Vorhofflimmern oder -flattern. Bei dieser Herzrhythmusstörung besteht ein erhöhtes Embolierisiko, das bei vielen Patienten durch Gerinnungshemmer gesenkt werden muss. Zweithäufigster Grund sind Thrombosen, meist der Beinvenen. Hier soll die Gerinnungshemmung in der Akutphase die weitere Ausdehnung der Thrombose und später ein Wiederauftreten (Rezidiv) verhindern.
Heparin
Zur medikamentösen Gerinnungshemmung in vivo können Heparin und Heparinoide eingesetzt werden. Es handelt sich um eine extrem stark negativ geladene Kette aus Zuckern, die sich an das schon erwähnte Protein Antithrombin heftet. Dieser Komplex bindet nun wirksamer die Faktoren Thrombin und Xa, die dadurch außer Kraft gesetzt werden: Die Gerinnungskaskade kommt zum Erliegen. Die Wirkung setzt nach intravenöser Gabe sofort ein. Heparin zur medikamentösen Verwendung wird üblicherweise aus tierischen Geweben gewonnen.
Cumarine
Eine weitere Möglichkeit sind sogenannte Vitamin-K-Antagonisten wie die Cumarinderivate Phenprocoumon und Warfarin. Vitamin K wird zur Synthese der meisten Gerinnungsfaktoren als Coenzym benötigt. Cumarin wirkt in der Leber und verhindert die Reduktion von Vitamin K (Phyllochinon). Dieses wirkt bei der γ-Carboxylierung der Gerinnungsfaktoren (II, VII, IX, X) mit und wird dabei selbst oxidiert (Abgabe von Elektronen). Ohne eine darauffolgende Reduktion (Aufnahme von Elektronen) bleibt Vitamin K funktionslos. Die Wirkung setzt zwar erst nach einer gewissen Zeit ein, dafür kann die Gabe oral erfolgen.
Thrombozytenaggregationshemmer
Acetylsalicylsäure kann in die Thrombozytenaggregation, also in die zelluläre Hämostase, eingreifen. Eine Cyclooxygenase (COX), die für die Synthese des Plättchenfaktors Thromboxan A2 benötigt wird, wird irreversibel durch Anheftung eines Essigsäure-Restes gehemmt. Ebenfalls auf die Aggregation der Blutplättchen wirkt Clopidogrel, das eine Hemmung der ADP-abhängigen Thrombozytenaktivierung durch eine irreversible Rezeptor-Blockierung bewirkt. Abciximab ist ein rekombinanter monoklonaler Antikörper, der das Glykoprotein IIb/IIIa der Thrombozyten blockiert und dadurch gleichfalls die Thrombozytenaggregation unterbindet. Denselben Angriffsort hat Tirofiban.
Fibrinolytika
Fibrinolytika aktivieren Plasminogen und fördern so die Auflösung von Thromben (Thrombolyse). Dies wird zur Therapie von Herzinfarkten, Lungenembolien, Beinvenenthrombosen, peripheren Verschlusskrankheiten und innerhalb eines vierstündigen Zeitfensters auch bei akuten Hirninfarkten genutzt. Während Wirkstoffe wie Streptokinase und Urokinase unspezifisch sowohl auf Fibrinogen als auch auf Fibrin wirken, weisen neuere Stoffe wie Alteplase (recombinant tissue type plasminogen activator, rt-PA) eine Selektivität für vernetztes Fibrin in Thromben auf, was systemische Nebenwirkungen, insbesondere Blutungen, vermindern soll. Die Anwendung der Fibrinolytika unterliegt einer strengen Indikationsstellung.
Hemmung in vitro
In vitro, z. B. in Blutröhrchen, kommen häufig EDTA und Citrat zum Einsatz, Chelatoren, die einen Komplex mit den zur Gerinnung nötigen Calcium-Kationen bilden. Eine Gerinnungshemmung mit Heparin ist in vitro ebenfalls möglich. Die Auswahl des Gerinnungshemmers erfolgt nach dem Gesichtspunkt, welche Untersuchung später mit dem ungerinnbar gemachten Blut geplant ist. Für Untersuchungen der Gerinnung selbst wird fast ausschließlich Citrat als Gerinnungshemmer verwendet, indem die Blutprobe im Verhältnis 9+1 mit einer 3,8%igen Natriumcitrat-Lösung verdünnt wird. Man verwendet dazu in der Regel industriell vorgefertigte Röhrchen, die bereits 0,3 ml Natriumcitratlösung enthalten und dann mit 2,7 ml Blut aufgefüllt werden. Für die Zuverlässigkeit der daraus erstellten Analysen ist es wichtig, dass dieses Mischungsverhältnis genau eingehalten und die Blutprobe sofort nach Gewinnung sorgfältig mit der Natriumcitrat-Lösung vermischt wird.
Medikamentöse Verstärkung der Hämostase
Es liegt nahe, die Hämostase auch in umgekehrter Richtung beeinflussen zu wollen und bei lebensbedrohlichen Blutungen Medikamente zu verabreichen, die zu einer verstärkten Hämostase führen. Die Entwicklung derartiger Medikamente, in der Fachsprache Hämostyptika genannt, war in der Vergangenheit – verglichen mit den die Hämostase hemmenden Medikamenten – von geringerem Erfolg gekrönt.
Für die medizinische Behandlung wichtig geworden sind hier vor allem Präparate, die einen angeborenen oder erworbenen Mangel von Gerinnungsfaktoren beheben, beispielsweise Faktor-VIII-Konzentrat bei Bluterkrankheit (Hämophilie A), Vitamin K und PPSB bei Blutungen unter Cumarintherapie oder gefrorenes Frischplasma bei disseminierter intravasaler Gerinnung. Bei einem ausgeprägten Mangel an Blutplättchen können diese in Form von Thrombozyten-Konzentraten ersetzt werden. Die Wirkung von Heparin kann durch Protamin aufgehoben werden.
Weiterhin kann die Hämostase verstärkt werden, indem der natürliche Gegenspieler der Gerinnung, die Fibrinolyse, gehemmt wird. Medikamente mit diesem Wirkmechanismus werden Antifibrinolytika genannt. Als Wirkstoffe kommen Tranexamsäure, para-Aminomethylbenzoesäure und ε-Aminocapronsäure zur Anwendung, das früher häufig verwendete Aprotinin wurde im November 2007 wegen erhöhter Sterblichkeit bei der Behandlung vom Markt genommen.[18]
Als ungeeignet, weil dessen Wirkungsweise sich im Bereich der Spekulation bewegte, erwies sich Butylalkohol im Präparat Hämostyptikum Revici.
Gerinnungsuntersuchungen
Das Messen der Blutgerinnung (Gerinnbarkeit, Koagulation, Koagulabilität) heißt Koagulometrie, entsprechende Geräte heißen Koagulometer. Das Ergebnis der Blutgerinnungsmessung ist der Gerinnungsstatus (Koagulogramm).[19][20]
| Abk. | Bezeichnung | Einheit | Normwert | Material | Aktivator | Monitoring von |
|---|---|---|---|---|---|---|
| TPZ, PT | Thromboplastinzeit, Prothrombin Time | Sekunden | 11-16 | Citratplasma nach Zentrifugation | Gewebethrombokinase = Tissue-Faktor = Thromboplastin = Faktor III | Zeitbeginn des extrinsischen Gerinnungssystems, Therapie mit Vitamin-K-Antagonisten |
| Quick | entspricht TPZ, PT im Vergleich zu Normplasma | Prozent | 70-125 % | Citratplasma nach Zentrifugation | s. o. | s. o. |
| INR | entspricht standardisierter TPZ, PT im Vergleich zu Normplasma | 0,8 – 1,2 | Citratplasma nach Zentrifugation | s. o. | s. o. | |
| aPTT | (activated) Partial Thromboplastin Time | Sekunden | 20-38 | Citratplasma nach Zentrifugation | Phospholipide (veraltet auch: partielles Thromboplastin oder Plättchenfaktor 3, ein proteinfreier Phospholipidextrakt), und eine oberflächenaktive Substanz (z. B. Kaolin) | Zeitbeginn des intrinsischen Gerinnungssystems, Heparintherapie |
| ACT | Activated Coagulation Time, Kaolin Clotting Time | Sekunden | 100-130 | Vollblut | oberflächenaktive Substanz (z. B. Kaolin) | Zeitbeginn des intrinsischen Gerinnungssystems, Heparintherapie, Messung patientennah aus Vollblut möglich z. B. bei HLM oder ECMO |
| PTZ, TZ | Plasmathrombinzeit, Thrombinzeit | Sekunden | 20-38 | Citratplasma nach Zentrifugation | Thrombin | Zeitbeginn der gemeinsamen Endstrecke des Gerinnungssystems, Heparintherapie |
| Multiplate® ASPI | kommerzielle Thrombozytenimpedanzaggregrometrie | Fläche unter der Kurve | >40 | Hirudinvollblut | Arachidonsäure (als Substrat für die COX zur Herstellung von Thromboxan A2) | Thrombozytenfunktion, COX-Hemmer: ASS (z. B. Aspirin), NSAR |
| Multiplate® ADP | kommerzielle Thrombozytenimpedanzaggregrometrie | Fläche unter der Kurve | >40 | Hirudinvollblut | ADP | Thrombozytenfunktion, ADP-Rezeptor-Antagonisten: Clopidogrel, PrasugrelThrombozytenfunktion |
| Multiplate® TRAP | kommerzielle Thrombozytenimpedanzaggregrometrie | Fläche unter der Kurve | >40 | Hirudinvollblut | Thrombin Receptor Activating Peptide (TRAP-6) | Thrombozytenfunktion, Glycoprotein IIb/IIIa-Antagonisten, mechanischer Thrombozytendefekt |
| ROTEM® EXTEM | kommerzielle Thrombelastometrie
CT=Clotting Time CFT=Clot Formation TIme MCF=Maximum Clot Firmness ML=Maximum Lysis |
Citratvollblut | Gewebethrombokinase = Tissue-Faktor = Thromboplastin = Faktor III | Zeitbeginn des extrinsischen Gerinnungssystems, Gerinnselfestigkeit, Gerinnselbestandsdauer, Therapie mit Vitamin-K-Antagonisten | ||
| ROTEM®
INTEM |
s. o. | Citratvollblut | Partielles Thromboplastin-Phospholipid aus Kaninchenhirn | Zeitbeginn des intrinsischen Gerinnungssystems, Gerinnselfestigkeit, Gerinnselbestandsdauer, Heparintherapie | ||
| ROTEM® HEPTEM | s. o. | Citratvollblut | Partielles Thromboplastin-Phospholipid aus Kaninchenhirn
+ Heparinase zum Beenden von Heparineffekt |
Zeitbeginn des intrinsischen Gerinnungssystems, Gerinnselfestigkeit, Gerinnselbestandsdauer,
nach Aufhebung der Heparinwirkung | ||
| ROTEM® FIBTEM | s. o. | Citratvollblut | Gewebethrombokinase = Tissue-Faktor = Thromboplastin = Faktor III
+ Cytochalasin D zur Thrombozytenhemmung |
Zeitbeginn des extrinsischen Gerinnungssystems, Gerinnselfestigkeit, Gerinnselbestandsdauer OHNE Thrombozyteneffekt d. h. der isolierte Fibrinogeneffekt zeigt sich | ||
| ROTEM®
APTEM |
s. o. | Citratvollblut | Gewebethrombokinase = Tissue-Faktor = Thromboplastin = Faktor III
+ Aprotinin zur Hemmung einer Hyperfibrinolyse |
insbesondere Gerinnselbestandsdauer, die durch eine Hyperfibrinolyse verkürzt wird |

Zur Messung der Gerinnungsfähigkeit des Blutes dienen in der labormedizinischen Diagnostik
- der Quick-Wert zur selektiven Funktionsbestimmung des exogenen Systems durch Zugabe von Tissue-Faktor und Ca2+ zur Blutprobe und anschließender Bestimmung der Gerinnungszeit im Vergleich zu Normalblut, beispielsweise bei einer Cumarintherapie, sowie die daraus abgeleitete INR (International Normalized Ratio), die den Quick-Test zunehmend ersetzt. Die INR bietet eine bessere Vergleichbarkeit zwischen verschiedenen Laboratorien als der Quick-Wert. Allerdings sind beide Werte bei einer Hämophilie normal.
- die PTT (Partial Thromboplastine Time) zur selektiven Funktionsbestimmung des endogenen Systems und des gemeinsamen Weges der Blutgerinnung.[21] Dieser Wert liegt bei einer Hämophilie über dem Standardwert von ca. 30 Sekunden.
Diese Untersuchungen werden als Globalteste der Gerinnung bezeichnet. Sie können nur eine reduzierte Gerinnung (Blutungsrisiko) erkennen und zur Überwachung einer Behandlung mit gerinnungshemmenden Medikamenten wie beispielsweise Marcumar dienen, nicht jedoch ein Zuviel (Thrombophilie). Weitere, seltener eingesetzte Tests zur Messung der Gerinnungsfähigkeit des Blutes sind Thrombinzeit und die funktionelle Fibrinogenbestimmung nach Clauss.
Der Aktivierungszustand des Gerinnungssystems im gesamten Körper kann durch die Messung der D-Dimere (Fibrinspaltprodukte) bestimmt werden. So können bestimmte zum Zeitpunkt der Blutentnahme vorhandene Krankheitszustände, die mit einer Aktivierung der plasmatischen Gerinnung einhergehen, erkannt werden (Thrombosen, Lungenembolien, disseminierte intravasale Gerinnung und Heparin-induzierte Thrombozytopenie Typ II). Eine Unterscheidung zwischen verschiedenen möglichen Ursachen einer Gerinnungsaktivierung sowie eine zuverlässige Einschätzung eines zukünftigen Risikos (Thrombophilie) ist durch die Bestimmung der D-Dimere nicht möglich. Ein geeigneter Suchtest für die Thrombophilie existiert zurzeit nicht, vielmehr müssen bei entsprechendem Verdacht alle möglichen Ursachen einzeln ausgeschlossen werden.
Eine Beurteilung des Quick-Werts und der PTT im Zusammenhang mit einer Blutungsneigung sollte immer eine eingehende Blutungsanamnese, die Zahl und ggf. auch die Funktion der Blutplättchen (Thrombozyten) mit einbeziehen. Die zelluläre Hämostase ist dabei wesentlich schwieriger einzuschätzen als die plasmatische. Einfach und zuverlässig bestimmbar ist nur die Zahl der Blutplättchen, nicht aber deren Funktion. Die für diesen Zweck vorgesehenen Tests sind entweder unzuverlässig (Blutungszeit) oder aufwändig und daher nicht überall verfügbar (Thrombelastogramm, Platelet Function Analyzer).
Vor Operationen wird auch bei Patienten, die keine gerinnungshemmenden Medikamente einnehmen, häufig eine grobe Einschätzung der Gerinnungssituation anhand dieser drei Parameter (Quick, PTT und Thrombozytenzahl) vorgenommen, um nicht-medikamentös bedingte Hämostasestörungen festzustellen. Diese Praxis ist mittlerweile in Expertenkreisen umstritten, da hier nur rund 13 % der Hämostasestörungen erkannt werden[22] und bei Ärzten ein Gefühl falscher Sicherheit erzeugt wird. Die epidemiologisch betrachtet häufigsten Gerinnungsstörungen, die die Thrombozytenfunktion bzw. den Von-Willebrand-Faktor betreffen, werden durch die drei Standardtests nicht erfasst, so dass die Durchführung nur noch bei positiver Blutungsanamnese empfohlen wird.[23][24] Andere Autoren halten dies wiederum für fahrlässig und empfehlen auch bei negativer Blutungsanamnese die routinemäßige präoperative Bestimmung von Thrombozytenzahl, aktivierter partieller Thromboplastinzeit (aPTT), Quick-Wert und Fibrinogen,[25] so dass weitere Studien hierzu nötig erscheinen.
Arterielles Blut gerinnt schneller als venöses, was auf die Differenzen im Gasgehalt zurückzuführen ist. Die Gerinnung arteriellen Bluts kann durch Zuführung von Kohlensäure verlangsamt, die des Venenbluts aber durch Vermehrung seines Sauerstoffgehalts beschleunigt werden. Die Verschiedenheiten in der Temperatur der beiden Blutarten sind viel weniger regelmäßig, denn während in Organen mit sehr lebhaftem Stoffwechsel (etwa Drüsen und Muskeln) das abfließende Blut wärmer ist als das eintretende, zeigen Organe mit nur unbedeutenden Wärmebildungsvermögen (beispielsweise die äußere Haut) ein umgekehrtes Verhalten.
Bedeutung bei Krankheiten
Grundsätzlich kann das Gleichgewicht zwischen Hämostase und Fibrinolyse in beide Richtungen entgleisen: Eine verstärkte Gerinnung wird als Thrombophilie bezeichnet (die dabei entstehenden, Krankheit verursachenden Blutgerinnsel werden als Thrombus beziehungsweise Embolus bezeichnet), eine reduzierte Gerinnung hämorrhagische Diathese. Eine Blutungsneigung kann dabei auch als Folge einer zuvor stattgefundenen starken Gerinnungsaktivierung mit Verbrauch von Gerinnungsfaktoren entstehen.
Blutungsneigung
Die oben beschriebenen physiologischen Vorgänge der Hämostase nach einer Verletzung (Blutgefäße, zelluläre und plasmatische Hämostase) können in jeder Phase gestört sein, so dass eine Reihe von verschiedenen Störungen jeweils zu einer Blutungsneigung führen können. Wie die Hämostase selbst können ihre Störungen bereits im Bereich der Blutgefäße beginnen. Beispielsweise kann eine angeborene Fehlbildung der Blutgefäße, die zu deren Erweiterung führt und als Morbus Osler bezeichnet wird, Ursache einer verstärkten Blutungsneigung sein.
Die zelluläre Hämostase ist bei einem ausgeprägten Mangel an Blutplättchen (Thrombozytopenie) oder bei Funktionsstörungen der Blutplättchen beeinträchtigt. Letztere sind die häufigste Ursache für eine verstärkte Blutungsneigung. Sie können durch Medikamente bedingt sein (siehe Abschnitt Thrombozytenaggregationshemmer oben), die häufigste angeborene Störung der zellulären Hämostase (und zugleich das häufigste angeborene Blutungsleiden überhaupt) ist das Willebrand-Jürgens-Syndrom.
Auch das Fehlen vieler plasmatischer Gerinnungsfaktoren kann zu teilweise lebensbedrohlichen Krankheiten führen, zum Beispiel bei erblichen Krankheiten wie der Hämophilie. Diese betrifft am häufigsten den Gerinnungsfaktor VIII (Hämophilie A), seltener auch den Gerinnungsfaktor IX (Hämophilie B).
Neben angeborenen Formen der Blutungsneigung, die in der Regel durch genetische Defekte einzelner Komponenten der Blutstillung bedingt sind, gibt es auch erworbene Zustände, die zu einer verstärkten Blutungsneigung führen. Die plasmatische Gerinnung kann z. B. durch einen Vitamin-K-Mangel beeinträchtigt werden. Dadurch können die Gerinnungsfaktoren II, VII, IX und X in der Leber nicht mehr ausreichend carboxyliert werden, was zu einem funktionellen Mangel und in der Folge insbesondere bei frühgeborenen Säuglingen zu schweren Hirnblutungen führen kann. Da alle Gerinnungsfaktoren in der Leber produziert werden, kommt es im Rahmen schwerer Lebererkrankungen nahezu regelhaft zu einem Mangel an Gerinnungsfaktoren mit der Folge einer erhöhten Blutungsgefahr.
Eine disseminierte intravasale Koagulopathie ist eine lebensbedrohliche Erkrankung, bei der durch einen abnormal hohen Spiegel körpereigener Botenstoffe wie Histamin, Serotonin und Adrenalin eine übermäßig stark ablaufende Blutgerinnung stattfindet. Dabei kommt es zu einem hohen Verbrauch der plasmatischen Gerinnungsfaktoren, die vom Körper nicht ausreichend schnell ersetzt werden können. Man spricht daher auch von einer Verbrauchskoagulopathie.
Bei Patienten mit einer Blutgerinnungsstörung können vor chirurgischen Eingriffen prophylaktische Plasmatransfusionen verwendet werden. Bezüglich der Wirkung von prophylaktischen Plasmatransfusionen vor invasiven Eingriffen bei Patienten ohne angeborene Blutgerinnungsstörungen ist die Evidenz für die Gesamtmortalität, für schwere Blutungen, für die Anzahl der Transfusionen pro Patient, für die Anzahl der Patienten, die eine Transfusion benötigen, und für transfusionsbedingte Komplikationen sehr ungewiss. Unterschiedliche Transfusionstrigger für gefrorenes Frischplasma (FFP) ermöglichen eventuell eine Verringerung der Anzahl der Personen, die eine solche Transfusion benötigen.[26]
Thrombosen und Embolien
Die Thrombose ist eine Gefäßerkrankung, bei der sich ein Blutgerinnsel (Thrombus) in einem Gefäß bildet. Ursachen dafür können in Schäden der Gefäßwand und generell in einem verminderten Blutdurchstrom gefunden werden. Doch auch Gerinnungsstörungen spielen hier eine große Rolle: So kann eine erbliche oder medikamentös herbeigeführte erhöhte Gerinnungsneigung schnell zu Thrombosen führen. Deshalb müssen beispielsweise auch bei langer Ruhigstellung der Beine Gegenmaßnahmen wie Medizinische Thromboseprophylaxestrümpfe (MTPS), Intermittierende pneumatische Kompression ergriffen, oder gerinnungshemmende Mittel wie Heparin oder Phenprocoumon gegeben werden.
Eine Embolie ist ein Thrombus, der von seinem Entstehungsort fortgeschwemmt wurde. Das kann zu schweren Komplikationen bis zum Hirninfarkt führen.
Thrombophilie
Es gibt eine Vielzahl an angeborenen und erworbenen Krankheiten, bei denen eine erhöhte Gerinnungsneigung besteht. Alle haben gemeinsam, dass es vermehrt zu Gefäßverschlüssen wie Thrombosen und Embolien kommt. Bei manchen Erkrankungen ist das Hochdrucksystem der Schlagadern (Arterien) stärker betroffen, bei anderen das Niederdrucksystem der Venen. Die häufigsten und wichtigsten Thrombophilien sind:
- Faktor-V-Leiden (zu mehr als 90 Prozent Ursache der APC-Resistenz)
- Prothrombinmutation G20210A
- Antiphospholipid-Syndrom
- Mangel an Inhibitoren der Gerinnung, insbesondere Protein-C-Mangel, Protein-S-Mangel und Antithrombin-Mangel
- erhöhtes Homocystein
- erhöhter Gerinnungsfaktor VIII.
Eine Sonderform der Thrombophilie kann im Rahmen der Behandlung mit dem gerinnungshemmenden Medikament Heparin auftreten. Durch dieses Medikament werden in einigen Fällen paradoxerweise die Blutplättchen aktiviert, so dass diese verklumpen und die Gerinnungskaskade in Gang setzen. Dies kann zu schweren Thrombosen im gesamten Körper führen. Messbar ist dabei der Abfall der Zahl der Blutplättchen, daher wird das Krankheitsbild als Heparin-induzierte Thrombozytopenie (Typ II) bezeichnet.
Siehe auch
- Blutstillung für weitere Methoden der medizinischen Blutstillung
Literatur
- Joachim Rassow, Karin Hauser, Roland Netzker: Biochemie. 1. Auflage. Thieme, Stuttgart 2006, ISBN 3-13-125351-7.
- Werner Müller-Esterl: Biochemie. Eine Einführung für Mediziner und Naturwissenschaftler. 1. Auflage. Spektrum Akademischer Verlag, Frankfurt 2004, ISBN 3-8274-0534-3.
- Roland Scholz: Medizinische Biochemie. 1. Auflage. Kap.11/12 : Biotransformation: Fremdstoffe, Häm, Cholesterin. Blutgerinnung und Fibrinolyse. Zuckerschwerdt, München 2003, ISBN 3-88603-822-X.
- Robert F. Schmidt, Florian Lang, Gerhard Thews: Physiologie des Menschen. 29. Auflage. Springer, Berlin 2004, ISBN 3-540-21882-3.
- Monika Barthels, Mario von Depka: Das Gerinnungskompendium. 1. Auflage. Thieme, Stuttgart 2003, ISBN 3-13-131751-5.
- Herbert A. Neumann: Das Gerinnungssystem: Physiologie und Pathophysiologie. Eine Einführung. 1. Auflage. ABW Wissenschaftsverlag, Berlin 2007, ISBN 3-936072-66-3.
- Samuel C. Harvey: The history of hemostasis. In: Annales of medical history. Neue Folge, 1, 1929, S. 127–134.
Weblinks
- Axel W. Bauer, Kerstin Mall: Hämostase, Thrombose und Embolie. Historische Konzepte zur Physiologie der Blutgerinnung (Memento vom 4. März 2010 im Internet Archive), Uni Heidelberg. Veröffentlicht in Hämostaseologie. 15 (1995) 92–99.
- Blood Coagulation (englisch)
Einzelnachweise
- Gerd Herold: Innere Medizin. Eigenverlag, Köln 2018, S. 135.
- Ulrich Weber: Biologie. Gesamtband Oberstufe, Cornelsen, Berlin 2001, ISBN 3-464-04279-0, S. 153.
- J. Staubesand: Bau und Funktion der Blutgefäße. In: Benninghoff Anatomie. 15. Auflage. Urban & Springer, München 1994.
- Robert F. Schmidt, Florian Lang, Gerhard Thews: Physiologie des Menschen. 29. Auflage. Springer, Berlin 2004, ISBN 3-540-21882-3, S. 524.
- Blood coagulation (en)
- Rainer Klinke, Hans-Christian Pape, Stefan Silbernagl (Hrsg.): Lehrbuch der Physiologie. 5. Auflage. Thieme, Stuttgart 2005, ISBN 3-13-796003-7; S. 246 f.
- Deetjen, Speckmann, Hescheler: Physiologie. 4. Auflage. Urban & Fischer, München 2006, ISBN 3-437-44440-9, S. 366.
- Earl W. Davie, Kazuo Fujikawa, Walter Kisiel: The coagulation cascade: initiation, maintenance, and regulation. In: Biochemistry, 1991, 30 (43), 10363–10370.
- Joachim Rassow, Karin Hauser, Roland Netzker: Biochemie. 1. Auflage. Thieme, Stuttgart 2006, ISBN 3-13-125351-7, S. 742.
- A Cell-based Model of Hemostasis, Maureane Hoffman, Dougald M. Monroe III, Thromb Haemost 2001; 85: 958–65 © 2001 Schattauer GmbH, Stuttgart, PMID 11434702.
- Die zentrale Rolle der Thrombozyten im neuen Verständnis der Hämostase, K. Jurk, B. E. Kehrel, Hämostaseologie 2005; 25: 39-49 Schattauer GmbH, Stuttgart.
- F. Josso, O. Prou-Wartelle: Interaction of tissue factor and factor VII at the earliest phase of coagulation. In: Thrombosis et diathesis haemorrhagica. Supplementum. Band 17, 1965, ISSN 0375-9997, S. 35–44, PMID 5874847.
- Wolf-Dieter Müller-Jahncke, Christoph Friedrich, Ulrich Meyer: Arzneimittelgeschichte. 2., überarb. und erw. Auflage. Wiss. Verl.-Ges, Stuttgart 2005, ISBN 978-3-8047-2113-5, S. 116.
- Axel W. Bauer, Kerstin Mall: Hämostase, Thrombose und Embolie. Historische Konzepte zur Physiologie der Blutgerinnung (Memento vom 4. März 2010 im Internet Archive), Uni Heidelberg. Veröffentlicht in Hämostaseologie. 15 (1995) 92–99.
- R. J. Jenny, D. D. Pittman, J. J. Toole, R. W. Kriz, R. A. Aldape, R. M. Hewick, R. J. Kaufman, K. G. Mann: Complete cDNA and derived amino acid sequence of human factor V. In: Proc Natl Acad Sci U S A, 1987, 84, S. 4846–4850, PMID 3110773.
- R. A. Biggs, A. S. Douglas, R. G. MacFarlane, J. V. Dacie, W. R. Pittney, C. Merskey und J. R. O’Brien: Christmas disease, a condition previously mistaken for haemophilia. In: British Medical Journal, London, 1952, S. 1378–1382.
- Physiologie der Gerinnung (Memento vom 15. September 2011 im Internet Archive) Werlhof-Institut.
- FDA Pressemitteilung vom 5. November 2007.
- Günter Thiele (Hrsg.): Handlexikon der Medizin, Urban & Schwarzenberg, München / Wien / Baltimore ohne Jahr, Band II (F–K), S. 1328.
- Lexikon Medizin, 4. Auflage, Elsevier Verlag, München ohne Jahr [2005], ISBN 3-625-10768-6, S. 920.
- B. Luxembourg et al.: Basiswissen Gerinnungslabor. In: Deutsches Ärzteblatt 104, Ausgabe 21, 25. Mai 2007, Seite A-1489.
- J. Koscielny et al.: Präoperative Identifikation von Patienten mit (primären) Hämostasestörungen. In: Hamostaseologie, 2007 Aug,27(3), S. 177–184, PMID 17694224.
- G. Pfanner et al.: Präoperative Blutungsanamnese. In: Anaesthesist, 2007 Juni 56(6), S. 604–611. doi:10.1007/s00101-007-1182-0, PMID 17522829.
- C. Bidlingmaier et al.: Haemostatic testing prior to elective surgery in children? Not always! In: Hamostaseologie, 2009 Jan, 29(1), S. 64–67, PMID 19151849.
- F. W. Albert et al.: Laboranalytischer Ausschluss einer hämorrhagischen Diathese vor elektiven Eingriffen? Ja!. (Memento des Originals vom 27. April 2015 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis. In: Hamostaseologie, 2009 Jan, 29(1), S. 58–63.
- Jonathan Huber, Simon J. Stanworth, Carolyn Doree, Patricia M. Fortin, Marialena Trivella: Prophylactic plasma transfusion for patients without inherited bleeding disorders or anticoagulant use undergoing non-cardiac surgery or invasive procedures. In: Cochrane Database of Systematic Reviews. 28. November 2019, doi:10.1002/14651858.CD012745.pub2 (wiley.com [abgerufen am 25. August 2020]).