Stuart-Prower-Faktor
Stuart-Prower-Faktor (auch Thrombokinase oder Faktor X) ist ein an der Blutgerinnung beteiligtes Enzym. Deshalb wird es zur Gruppe der Gerinnungsfaktoren gezählt.
| Faktor Xa | ||
|---|---|---|
| ||
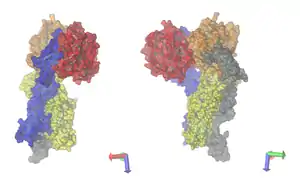
| Bänder-/Oberflächenmodell von Faktor Xa (blau/rot) mit Faktor VIIa (dunkelgrün/grau) und Thromboplastin (hellgrün) nach PDB 1NL8. | ||
|
Vorhandene Strukturdaten: s. UniProt | ||
| Eigenschaften des menschlichen Proteins | ||
| Masse/Länge Primärstruktur | 447 = 139+306 Aminosäuren | |
| Kofaktor | Faktor Va, Ca2+, Phospholipid | |
| Bezeichner | ||
| Gen-Namen | F10 ; FX; FXA | |
| Externe IDs | ||
| Enzymklassifikation | ||
| EC, Kategorie | 3.4.21.6, Serinprotease | |
| MEROPS | S01.216 | |
| Substrat | Arg-+-Thr und Arg-+-Ile in Prothrombin | |
| Produkte | Thrombin | |
| Vorkommen | ||
| Homologie-Familie | Trypsin | |
| Übergeordnetes Taxon | Lebewesen | |
| Orthologe | ||
| Mensch | Hausmaus | |
| Entrez | 2159 | 14058 |
| Ensembl | ENSG00000126218 | ENSMUSG00000031444 |
| UniProt | P00742 | O88947 |
| Refseq (mRNA) | NM_000504 | NM_001242368 |
| Refseq (Protein) | NP_000495 | NP_001229297 |
| Genlocus | Chr 13: 113.12 – 113.15 Mb | Chr 8: 13.04 – 13.06 Mb |
| PubMed-Suche | 2159 | 14058 |
Physiologie
Der Stuart-Prower-Faktor ist ein Gerinnungsfaktor des Menschen. Er wird in der Leber synthetisiert, dazu wird Vitamin K benötigt. Er gehört zur Gruppe der α-Globuline und besteht aus einer schweren und einer leichten Kette. Er hat eine Molekülmasse von 59 kDa und gehört zu den Serinproteasen, mit einer EC-Nummer von 3.4.21.6. Seine Halbwertszeit im Blut beträgt 40 bis 45 Stunden, seine Plasmakonzentration 7–10 mg/l.
Bei der Aktivierung des Stuart-Prower-Faktors wird eine Peptidbindung innerhalb der schweren Kette, die das aktive Zentrum trägt, gespalten. Dies kann auf zwei verschiedenen Wegen aktiviert werden. Beim extrinsischen Pfad, der eintritt, wenn Blutgefäße beschädigt werden, aktiviert ein Komplex aus aktiviertem Proconvertin (Faktor VIIa) und Gewebefaktor (Faktor III) den Stuart-Prower-Faktor (Faktor Xa). Im Gegensatz dazu wird er beim intrinsischen Pfad, der eintritt, falls Teile der Gerinnungskaskade auf negativ geladene Teilchen (wie zum Beispiel Kollagen unter dem Endothel) treffen, durch einen Komplex aus dem aktivierten Christmas-Faktor (Faktor IXa) und antihämophilem Globulin A (Faktor VIIIa) aktiviert. Außerdem kann er durch das Verdauungsenzym Trypsin und das Gift der Russel-Viper aktiviert werden.
Faktor Xa ist deshalb der erste Schritt der Gerinnungsphase, alles davor gehört zur Aktivierungsphase. Er spaltet Prothrombin (Faktor II) an zwei Stellen; zwischen einer Arg-Thr-Bindung und zwischen einer Arg-Ile-Bindung. Dies wandelt Prothrombin in Thrombin um. Für diesen Prozess benötigt der Stuart-Prower-Faktor aktiviertes Proaccelerin (Faktor Va) als Kofaktor.
Faktor Xa wird durch eine von Protein Z abhängige Serinproteinase inaktiviert. Die Affinität dieses Enzyms für Faktor Xa wird durch Protein Z vertausendfacht. Defektes Protein Z führt zu einer erhöhten Aktivität des Stuart-Prower-Faktors und einem Hang zur Bildung von Thrombosen.
Erkrankungen
Faktor-X-Defekte treten in Verbindung mit Nasenbluten (Epistaxis), Gelenksblutungen (Hämarthrose) und Blutverlust im Verdauungstrakt (gastrointestinale Blutungen) auf. Faktor-X-Defekte können angeboren oder erworben sein. Der angeborene Defekt ist selten (1:500.000). Das Gen des Stuart-Prower-Faktors liegt beim Menschen auf Chromosom 13, Genlocus q34.
Ein vermindertes Vorkommen des Stuart-Prower-Faktors kann bei verschiedenen Krankheiten erworben auftreten, wie z. B. Amyloidose. Ein Mangel an Vitamin K führt zur Bildung von inaktivem Faktor X. Bei einer Vitamin K Hemmung durch Warfarin Therapie oder ähnliche Antagonisten ist dieser Effekt zur Vermeidung von Thrombosen erwünscht.
Verwendung
Faktor X ist als Konzentrat in gefrorenem Blutplasma und im Prothrombin-Komplex enthalten. 2012 war das einzige zugelassene Faktor-X-Konzentrat ist Faktor X P Behring, hergestellt von CSL Behring.[1]
Ein hochreines, aus Plasma gewonnenes Faktor X-Konzentrat (pdFX) ist in den Vereinigten Staaten und in der Europäischen Union als Ersatztherapie für FXD zugelassen. Daten zur Verwendung von pdFX zur Prophylaxe der Blutung bei Kindern unter 12 Jahren wurden 2018 veröffentlicht.[2]
Andexanet alfa ist ein rekombinantes Analogon von Faktor Xa.
Forschungsgeschichte
Der Faktor-X-Mangel wurde von britischen (C. Hougie et al.), deutschen (M. Hörder) und schweizerischen (K.-D. Bachmann et al.) Forschern unabhängig voneinander entdeckt und in den Jahren 1957 und 1958 beschrieben.[3] Wie andere Gerinnungsfaktoren wurde der Faktor X nach jenen Patienten benannt, bei denen ein Mangel erstmals beschrieben wurde. Die Nomenklatur stammt von der schweizerischen Arbeitsgruppe um K.-D. Bachmann.[4]
Siehe auch
Weblinks
- Stuart-Prower-Faktor. In: Online Mendelian Inheritance in Man. (englisch)
- Factor X (Emedicine, engl.)
- Factor X Mangel (engl.)
Einzelnachweise
- Mark Brooker: Registry of Clotting Factor Concentrates. 9. Ausgabe, World Federation of Hemophilia, 2012.
- R. Liesner, C. Akanezi u. a.: Prophylactic treatment of bleeding episodes in children <12 years with moderate to severe hereditary factor X deficiency (FXD): Efficacy and safety of a high-purity plasma-derived factor X (pdFX) concentrate. In: Haemophilia. 24, 2018, S. 941, PMID 29707881. doi:10.1111/hae.13500.
- Ludwig Heilmeyer: Referat über die Habilitationsschrift von M.H. Hörder „Experimentelle und klinische Untersuchungen über den V. Blutgerinnungsfaktor“. 1959.
- Max-Hermann Hörder: Experimentelle und klinische Untersuchungen über den fünften Blutgerinnungsfaktor, Habilitationsschrift, 1959, S. 69 DNB 480023476.